







Introduction
Peganum harmala L., known as harmel, esfand, aspand, and wild or Syrian rue, is an untamed halonitrophilous perennial herb widely spread in Asia, Mediterranean Europe, Northern Africa, and America (Güemes and Sánchez-Gómez, 2015). It belongs to the Nitrariaceae family (previously belonged to Zygophyllaceae), which has three genera, namely Nitraria L., Peganum L., and Tetradiclis M. Bieberstein. It grows naturally in abandoned and non-cultivated regions in steppe, semidesert, and desert climates where there is no agricultural activity for a long time (Abbott et al., 2008). It is a resistant plant that can tolerate adverse ecological conditions and environmental cues, especially salinity, cold, and drought (Karam et al., 2016; Abbott et al., 2008). Being a drought-resistant and salt-tolerant species, P. harmala plays a crucial bionomic role in plant defense against disease and insects; water and soil conservation; maintaining biological diversity; and ecosystem restoration (Li et al., 2018). The resistance and wildness of P. harmala make it a biologically interesting plant for studying stress, evolution, and biodiversity (Zha et al., 2020).
P. harmala is an alkaloid plant traditionally used in the treatment of cardiovascular, gastrointestinal, nervous, and endocrine diseases; treatment of tumors; and pain relief, with antidiabetic and antiseptic activities (Miao et al., 2020). It contains alkaloids that primarily belong to beta-carbolines and quinazolines. It produces two major metabolite groups, namely H-metabolites such as harmine, harmaline, harmalol, and harman; and P-metabolites such as pegamine, peganol, and peganone (Miao et al., 2020). Its alkaloids possess a heterocyclic structure containing two nitrogen elements as their alkaloidal core. These two alkaloid groups are representative of the genus Peganum and make it a valuable multipurpose source of alkaloids because of a variety of activities such as antiseptic, intoxicant, antispasmodic, stimulant, antidepressant, aphrodisiac, diuretic, sedative, and narcotic. These effects have been reported for whole plant, seeds, flowers, inflorescences, nectar, and roots (Moloudizargari et al., 2013; Mahmoudian et al., 2002). Besides alkaloids, other metabolites such as fatty acids, alkanes, and essential oils such as hexadecanoic acid are abundant in P. harmala (Moussa and Almaghrabi, 2016). Using NMR-based metabolomic profiling, 24 compounds including species-specific alkaloids such as vasicine, vasicinone, harmine, and harmaline in the P. harmala metabolome have been detected (Li et al., 2018). Levels of some alkaloids significantly change depending on seasons; the vasicinone content is higher in December compared with May, October, and August, and the vasicine content is higher in May compared with the other studied months.
Medicinal plants are also used in plant disease control as their metabolites are protective agents against numerous organisms attacking plants. The total alkaloid extract of P. harmala is an antibacterial solution that has been used to affect bacteria in vivo and in vitro, including Ralstonia solanacearum Phylotype II, Erwinia amylovora, Pectobacterium carotovorum subsp. carotovorum, and Burkholderia gladioli (Shaheen and Issa, 2020). This study showed that the 300 μg/ml concentration of the P. harmala extract had the most remarkable effect on the studied bacteria without phytotoxicity, suggesting the application of P. harmala alkaloids as antibacterial agents.
Despite its potential, genomic and transcriptomic data of P. harmala are not sufficiently available. Genome and transcriptome studies help researchers and scientists understand the metabolite biosynthesis capacity of P. harmala to produce alkaloids and its adaptive responses to adverse environmental cues. However, some studies on molecular markers show the existence of genetic variations between P. harmala populations. El-Bakatoushi and Ahmed (2018) studied genetic diversity using inter-simple sequence repeats (ISSRs), polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP) of ribosomal DNA internal transcribed spacer (rDNA-ITS), PCR-SSCP (single strand conformation polymorphism) of rDNA-ITS, and simple sequence repeat (SSR) markers and reported that ITS-SSCP and ISSR markers were more informative than other markers in the detection of variations between different P. harmala populations (EL-Bakatoushi and Ahmed, 2018). In a gene expression profiling and comparative study between Arabidopsis thaliana and P. harmala, catalase genes cat1, cat2-3, and cat3 were overexpressed in response to salinity, showing their role in salinity tolerance of P. harmala (Karam et al., 2016). Another study reported 21 P. harmala accessions collected from different regions in Iran, which were categorized into three genotype groups based on their geographical distribution and principal coordinate analysis, suggesting the occurrence of genetic variations between native P. harmala plants of Iran (Zebarjadi et al., 2016). The genome size of P. harmala is 0.61–0.67 pg, and it has 24 chromosomes (Hajji et al., 2017). However, to the best of our knowledge, neither published transcriptome nor genome sequences of P. harmala are available.
The de novo assembly technique of genomes and transcriptomes for non-model plants is an efficient approach (Huang et al., 2016) by which genomes and transcriptomes of numerous plants have been generated for the first time. However, for vertebrate genomes, using one genome assembler solely might not be sufficient to assemble the whole genome and a percentage of genes and sequences will be lost (Rhie et al., 2021). Hence, the simultaneous use of different approaches and assemblers is appropriate for generating more accurate genome assemblies (Rhie et al., 2021). A combination of different approaches and state-of-the-art pipelines for de novo assembly and mapping genomes to ameliorate the assembly has been proven more practical for different plant species (Cerveau and Jackson, 2016; Lischer and Shimizu, 2017; Jazayeri, 2015). The genome assembly of Klebsiella pneumoniae by hybrid pipelines in which different assemblers such as Canu + pillon and Mini + pillon were used was created (Bayat et al., 2020). A combination of de novo assembly and mapping against reference genome, when a reference is available, results in a more accurate genome assembly (Visser et al., 2015). In a Ribonucleic Acid sequencing (RNA-seq) study, 17 individuals across five populations of the haplotype human genome were used in genome assembly. This study revealed 1842 breakpoint-resolved nonreference unique insertions that added 2.1 Mb content to the human genome (Wong et al., 2018). The computation time and calculation resources as well as the aim of the study are factors to be considered while conducting genomic and transcriptomic analyses. These factors should be considered while designing a suitable workflow (Cerveau and Jackson, 2016).
RNA-seq is one of the most used techniques in biology, with a growing rate of around 3000 times in 6 years (2008–2014) (Jazayeri et al., 2015). It has helped answer many biological questions and improve plant stress studies, growth and development analyses, and disease diagnosis (Vitoriano and Calixto, 2021; Marco-Puche et al., 2019). It also helps understand biological functions and mechanisms involved in plant adaptation, for example, as proven in a study of sweet potato where TF families including basic helix–loop–helix (bHLH), basic leucine zipper (bZIP), C2H2, C3H (Cys3His zinc finger), ethylene-responsive transcription factor (ERF), homo domain-leucine zipper, MYB (myeloblastosis), NAC (NAM (no apical meristem), ATAF1/2 (Arabidopsis transcription activation factor), CUC2 (cup-shaped cotyledon)), thiol-specific antioxidant, and WRKY domain (W, R, K, Y stand for amino acids tryptophan, arginine, lysine and tyrosine respectively) were disclosed involved in drought stress response (Arisha et al., 2020), and biodiversity as reported by nucleotide-binding site and leucine-rich repeats (NBS-LRR) resistance genes in walnut (Chakraborty et al., 2016). A study on rice leaf response to heat stress identified 17 143 and 2162 heat response genes using RNA-seq data analyses of differential gene expression and differential alternative splicing, respectively (Vitoriano and Calixto, 2021). In another study differentially expressed genes involved in oil palm drought stress response were revealed to screen tolerant and susceptible oil palm plants (Jazayeri, 2015).
This in silico research aimed to develop the first transcriptome report for P. harmala. A de novo assembled transcriptome, its annotation, and genes related to alkaloid biosynthesis and those involved in stress responses were reported. In addition, this report presented the details on regulatory and transcriptional elements of P. harmala obtained via transcriptomic analyses. The transcriptome generated can be used in subsequent studies on metabolite production, stress tolerance, and biodiversity.
Materials and methods
Samples and sequencing
The 1KPlant Project (Wickett et al., 2014) data of RNA sequencing for P. harmala that were used in the present study were obtained from the data deposited under the National Center for Biotechnology Information (NCBI) experiment number ERX2099528. Paired raw RNA reads were stored in the Sequence Read Archive (SAR) database under the NCBI accession number ERR2040471 as mentioned by the 1KPlant Project (Matasci et al., 2014). After downloading the sequencing data from the NCBI, raw 90-bp reads were assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads and bases and the likely contamination were eliminated by Trimmomatic (Bolger et al., 2014).
De novo transcriptome assembly
To ameliorate the assembly, a pipeline of de novo assembly was designed using two programs, namely Trinity (Haas et al., 2013) and SOAPdenovo-Trans (Luo et al., 2012). The clean reads were assembled to the contigs using Trinity with Group Pairs Distance 500, Path Reinforcement Distance 75, and other default parameters. SOAPdenovo-Trans was used with the default parameters. After merging de novo assembled outputs generated using the two assemblers, the transcripts were further processed using Cluster Database at High Identity with Tolerance (CD-HIT-EST) (Fu et al., 2012) at the 90% identity cutoff to reduce redundancy and remove identical fragments. AssemblyPostProcessor under the Galaxy Server (Goecks et al., 2010) was used to postprocess the assembled transcripts into putative coding sequences and their respective amino acid translations with open reading frames (ORFs) based on the Transdecoder method of the PlantTribes collection of automated modular analysis pipelines (Wall et al., 2008), considering default parameters as the basic option in the Galaxy Server. The transcriptome obtained using the postprocess step was used for subsequent annotation analyses.
Transcriptome assembly quality evaluation
To verify the accuracy of the de novo assembly, quality metrics including length- and annotation-based parameters were compared. For contig and transcript information, parameters such as N50 length, N90 length, total contig number, average and maximum contig length, GC (Guanine, Cytosine) and AT (Adenine, Thymine) content, and A/T and G/C ratio were assessed for each assembled transcriptome and the merged transcriptome using the Next Generation Sequencing Quality Control (NGS QC) Toolkit (Patel and Jain, 2012). In addition, raw reads were aligned back to the assembled transcriptomes using Hisat2 (Kim et al., 2015) with default parameters, and the percentage of the mapped back reads to transcripts was evaluated. On the other hand, Benchmarking Universal Single-Copy Orthologs (BUSCO) was used to assess the transcriptome assembly and annotation completeness using orthologs against Embryophyta obd10 (Waterhouse et al., 2018).
Annotation of transcript models and protein homology
To recognize the homologous protein, all transcript sequences were aligned against the NR (nonredundant), UniProt (SwissProt), plant TrEMBL, The Arabidopsis Information Resource (Araport11), and eukaryotic orthologous groups (KOG) protein databases using the NCBI-BLAST software (V2.2.30+) with an e-value threshold of 10E-10. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was conducted using the KEGG Automatic Annotation Server (KAAS; http://www.genome.jp/kegg/kaas/) with the single directional best hit method for KEGG orthology (KO) assignments and pathway mapping. To ameliorate the annotation for protein, GO, and KEGG, the transcriptome was subjected to the eggNOG-Mapper, which is a web-based tool based on the eggNOG 4.5 framework. It combines the public domains to retrieve more accurate information (Huerta-Cepas et al., 2017; Huerta-Cepas et al., 2016), and in the present study, it was used to generate graphs and pertinent outputs using the extensible markup language (XML) output files of blastx searches.
Protein sequences of Citrus sinensis with the accession number GCF_000317415.1 (Xu et al., 2013), Citrus clementina with the accession number GCA_0004 93195.1 (Wu et al., 2014), and Citrus unshiu with the accession number GCA_002897195.1 (Shimizu et al., 2017) – species closely related to P. harmala belonging to the same order Sapindales – were downloaded from the NCBI Genome database and utilized for homology search. The whole transcripts were annotated against three Citrus species protein models using blastx. To carry out a homology comparison with other alkaloid plants, the transcript sequences were searched against Catharanthus roseus (available at http://medicinalplantgenomics.msu.edu/pub/data/MPGR/Catharanthus_roseus/) (Kellner et al., 2015), Nicotiana tabacum with the accession number GCF_000715135.1 (Sierro et al., 2014), Eschscholzia californica with the accession number BEHA01000001-BEHA01053253 (available at http://eschscholzia.kazusa.or.jp/) (Hori et al., 2018), and Papaver somniferum with the accession number GCA_00357 3695.1 (Guo et al., 2018) using blastx. The blast hits were filtered by the e-value cutoff of 10E-10. However, considering the stress-responsive attributes, two plant models – Arabidopsis halleri and Eutrema salsugineum protein datasets were used as they are established as plant models for metal stress responses and accumulation (A. halleri ) (Briskine et al., 2017) and for salinity, cold, and water-deficit responses (E. salsugineum ) (Yang et al., 2013). Moreover, they belong to the Brassicaceae family and the Brassicales order, which is close to Sapindales, and their comparison can reveal the likely relationship between them. Phytozome V12 was used to download the A. halleri protein dataset. The protein model dataset of E. salsugineum was downloaded from the NCBI, which was available under the accession number GCF_000478725.1, and compared with the P. harmala transcriptome using blastx with the cutoff of 10E-10.
To retrieve GO terms, the transcripts were annotated against the plant database of AgBase using the GOanna tool (McCarthy et al., 2006). GOanna is a tool from the AgBase collection (http://agbase.arizona.edu/cgi-bin/tools/GOanna.cgi) that allows to quickly add more GO annotations to data by transferring GO annotations based on sequence homology. GOSlimViewer was used to analyze the GO terms. The Interactive Tree Of Life (iTOL, https://itol.embl.de/) was used to generate the phylogenetic tree for P. harmala and some plant species of Sapindales, Malvales, and Brassicale. The online tool iTOL was used in the display, annotation, and management of phylogenetic trees (Letunic and Bork, 2016).
Alkaloid-related gene discovery
For alkaloid annotation, the public domains NCBI and Phytozome were used to compile alkaloid-related genes. The term “alkaloid” was searched in the NCBI Protein database, and “genome,” “chromosome,” and “scaffold” sequences were removed using the “NOT” delimiter. In addition, the term “Embryophyta” was used as a search criterion to limit the search to plant sequences. On the other hand, the Phytozome database was searched for alkaloid-related genes using the keyword “alkaloid”. The two compiled sequences were combined and clustered to remove redundancy using CD-HIT with a 90% similarity cutoff (Suppl. file 1 – https://data.mendeley.com/datasets/bxcx9jhzdt). The transcripts were subjected to a blastx-based analysis to find P. harmala alkaloid-related orthologs using the e-value cutoff of 10E-10. In addition, the alkaloid-related transcripts of P. harmala were imported to the GOanna tool of AgBase (McCarthy et al., 2006) to annotate them and search their pertinent GO terms.
In addition, because of the importance of cytochrome P450 in alkaloid biosynthesis, pertinent sequences of the cytochrome P450 family were downloaded from the Arabidopsis site (www.arabidopsis.org). The P. harmala transcriptome was blasted against all the downloaded cytochrome P450 proteins using the e-value cutoff of 10E-10.
Stress-responsive gene annotation
To determine stress-related transcripts with more concrete data, two different approaches were used. First, the annotated transcriptome of P. harmala was mined using the GO terms related to stress, including “stress,” “response to abiotic stimulus,” and “response to biotic stimulus.” Second, the SwissProt database was searched using syntax keywords “name: stress database: (type: ensemblplants) AND reviewed: yes” to shortlist stress-related proteins. The assembled transcripts were blasted against the stress proteins extracted from the SwissProt database using blastx with the e-value cutoff of 10E-10. Finally, the two lists of the P. harmala transcripts generated from GO term analysis and blast against SwisProt stress genes were compared, and the common transcripts were designated as stress-related genes evidenced by both datasets. These stress-related transcripts were analyzed using PlantTFcat, Goanna, and KAAS to scrutinize the pathways involved.
Identification of TFs and gene regulators
Using all transcript sequences of the newly generated P. harmala transcriptome, TFs and chromatin regulators as well as other regulatory and transcriptional elements were predicted and classified using PlantTFcat (Dai et al., 2013). PlantTFcat categorizes the sequences based on TF families and protein domains of the InterPro database.
Prediction of SSR markers
SSRs of the newly generated P. harmala transcriptome were identified using the Perl script MISA (Micro SAtellite identification tool, http://pgrc.ipk-gatersleben.de/misa). The MISA parameters were set to identify the SSRs of di-, tri-, tetra-, penta-, and hexa-nucleotides with the minimum number of repetitions of 6, 5, 4, 4, and 4, respectively. For compound SSRs (distinct and adjacent SSRs), a distance of 100 bp between two SSRs was adjusted. The primers for the transcripts possessing SSRs were generated using BatchPrimer3 (You et al., 2008). The transcript sequences of SSRs were submitted to BatchPrimers3, and the default settings for generic primers were applied as follows: product size of 500–1000 base pairs (bp), Max 3′ Stability 9.0, Max Mispriming 12.00, Pair Max. Mispriming 24.00, Primer size 18–27 nt (nucleotide), Primer Tm (temperature) 57.0–63.0°C, Max Tm Difference 10.00°C, Primer GC% 20–80%, Max Self Complementarity 8.00, Max 3′ Self Complementarity 3.00, and Max #N′s 0. The FastPCR software (Kalendar et al., 2017) was used to verify the accuracy of SSR markers using in silico PCR and unique PCR methods. The primers generated by using Batch Primer3 were inputted to FastPCR.
MicroRNA prediction
To identify microRNA families, the genomic loci encoding miRNAs in the P. harmala transcriptome were computationally predicted using psRNATarget (https://www.zhaolab.org/psRNATarget/). psRNATarget uses two methods to identify the plant sRNA targets: 1) complementary matching analysis between the sRNA sequence and the target mRNA sequence using a predefined scoring schema and 2) target site accessibility evaluation (Dai et al., 2018). Using psRNATarget, the microRNA targets of P. harmala were annotated to identify their regulatory attribute based on the target gene function. On the other hand, mature miRNA sequences of Citrus species from miRBase, namely C. clementina, C. reticulata, C. sinensis, and C. trifoliata, were mapped to the transcript sequences of P. harmala using psRNATarget. For stress-related microRNA, microRNA of E. salsugineum (a plant model for drought, cold, and salinity stress studies) (Yang et al., 2013) was aligned against the P. harmala transcriptome.
Results and discussion
P. harmala de novo transcriptome assembly
Four transcriptomes were generated, including one using Trinity, one using SOAPdenovo-Trans, one by merging transcriptomes generated by Trinity and SOAPdenovo-Trans, and finally, the integrated transcriptome that was a processed transcriptome of the other three generated transcriptomes (Table 1). A comparison of the four transcriptome assemblies showed that the merged and postprocessed transcriptomes were better structured than the Trinity and SOAPdenovo-Trans assemblies, thus confirming the effectiveness of the merging method. de novo assembly was performed on 12 528 661 paired-end sequence reads of P. harmala containing 2.3 Gbp, where the mean sequence length was 90 bp and the GC content was 45%. The transcriptome de novo assembled using Trinity contained 48 638 transcripts, and the SOAPdenovo-Trans transcriptome assembly generated 47 330 transcripts. Following the pipeline designed to merge the de novo assemblies, the generated transcriptome contained 67 211 transcripts after removing redundancy and clustering. The de novo assemblies were evaluated using the following parameters: total sequences and bases, sequence lengths, N50 and N90 length, A + T and G + C values, percentage of back-mapped assembly, and N count (Table 1). These values can be used to compare assemblers and to show how different methods and assemblers can be efficient, but it is not recommended to evaluate the accuracy (Ekblom and Wolf, 2014), which should be done using the back-mapping method. In general, Trinity showed better assembly characteristics than SOAPdenovo-Trans (Table 1). The quality control values for the de novo assembled transcriptomes were close between the created transcriptomes since we used the default parameters for both assemblers. However, the integrated transcriptome showed closer quality average values and higher coverage than each assembler individually. SOAPdenovo-Trans was previously used in the 1KPlant project to de novo assemble transcriptomes with default parameters, with the exception of the use of 29-mers in de Bruijn graph construction (Wickett et al., 2014).
Table 1
Transcriptome assembly quality evaluation metrics for four different transcriptome assembly approaches used in this study on Peganum harmala ; the four transcriptome assembly approaches, Trinity, SOAPdenovo-Trans, merged Trinity and SOAPdenovo-Trans, and integrated transcriptome, were analyzed using quality and annotation parameters
To corroborate assembly quality and completeness by each assembler and the merged one and to compare the assemblies, sequencing reads were back-mapped to the assembled transcriptome, stating the read coverage quantity to construct the assembly. Trinity using the single k-mer length convened slightly (around 2.3%) more sequencing reads to construct the assembly, compared with SOAPdenovo-Trans. Nevertheless, the merged assembly showed more back-mapped reads and was almost similar to that of Trinity. In addition, the merged transcriptome assembly showed the highest percentage of mapped reads in pairs and the lowest aligned discordant pair reads (Table 1). The assembly completeness can be verified using the percentage of reads back-mapped to the assembled transcripts, which shows the mapped read ratio between the input and the output of the assembler (Moreton et al., 2014). In the present study, the assembly procedure confirmed that merging different generated transcriptomes using different assemblers can fructify assembly, especially for non-model plants and when no reference is available, as reported for species such as oil palm (Cerveau and Jackson, 2016; Jazayeri, 2015). The pipeline followed in the present study is one of the PlantTribes automated modular analysis pipelines for comparative and evolutionary analyses of genome-scale gene families and transcriptomes (Wall et al., 2008).
The postprocess assembly step outputted 42 528 coding sequence transcripts as representative of P. harmala (Suppl. file 2 – https://data.mendeley.com/datasets/bxcx9jhzdt). Additionally, 30 281 coding protein sequences with ORFs were predicted using AssemblyPost Processor. AssemblyPostProcessor generated longer transcripts that seemed to be complete coding sequences by which the assembly can be more accurate compared with the merged assembly (Wall et al., 2008). It is generally used to ameliorate the assembly and transcripts (Wall et al., 2008), as also confirmed in the present analysis. The hallmarks of the postprocessed transcriptome, which contained 42 528 transcripts, were highly similar to the closely related C. sinensis and Pistacia vera species; then, the postprocessed generated transcriptome was presented as the representative for P. harmala.
Besides statistical parameters such as the N50 value and the number of transcripts of more than 1 kb size, the generated transcriptome was evaluated for the presence or absence of conserved orthologous genes and transcriptome completeness, which is a complementary assessment. For this analysis, the BUSCO library of Embryophyta orthologous genes was used. It includes representative single-copy orthologs belonging to the plant kingdom and provides important validation of the depth and completeness of the assembly (Waterhouse et al., 2018; Cerveau and Jackson, 2016). Among the transcriptomes assembled in the present study, the merged transcriptome of Trinity and SOAPdenovo-Trans showed 82.1% of transcript completeness, followed by the postprocessed integrated transcriptome with 81.2% and then the transcriptome assembled by Trinity and SOAPdenovo-Trans (Table 1). According to the BUSCO results, the merged and postprocessed integrated transcriptomes showed 78 and 84 missing hits, respectively, whereas Trinity and SOAPdenovo-Trans assemblies showed 87 and 89 missing hits, respectively. However, the higher number of complete and duplicated transcripts showed better depth and completeness of the assembly. This can validate the assembly as it compares the transcripts with the previously recognized single-copy orthologs as the model.
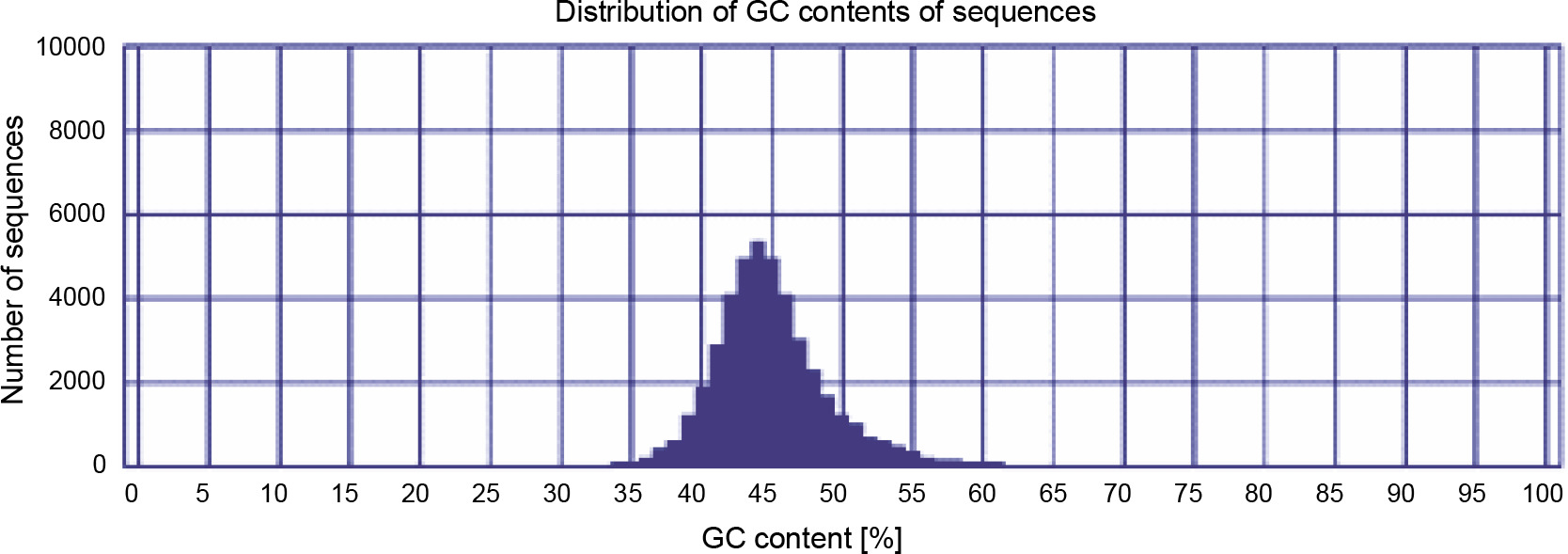
The GC content can elucidate biodiversity and evolutionary relationship between closely related plant species and is useful to compare different regions of a genome such as introns and exons (Singh et al., 2016). If the comparison of a newly assembled genome or transcriptome with the genome/transcriptome of other closely related species results in a higher similarity and closeness, the genome/transcriptome is well assembled. The GC content (around 42%) for three assemblies was very close to each other; however, the integrated postprocessed assembly had a GC content of around 44% (Fig. 1), which is highly similar to those of the closest species Citrus (Xiong et al., 2017) and P. vera (Moazzam Jazi et al., 2017). Eventually, based on different quality assessments, the integrated postprocessed transcriptome was found to be a better representative for P. harmala, and it was used for subsequent analyses.
Functional annotation of P. harmala transcriptome
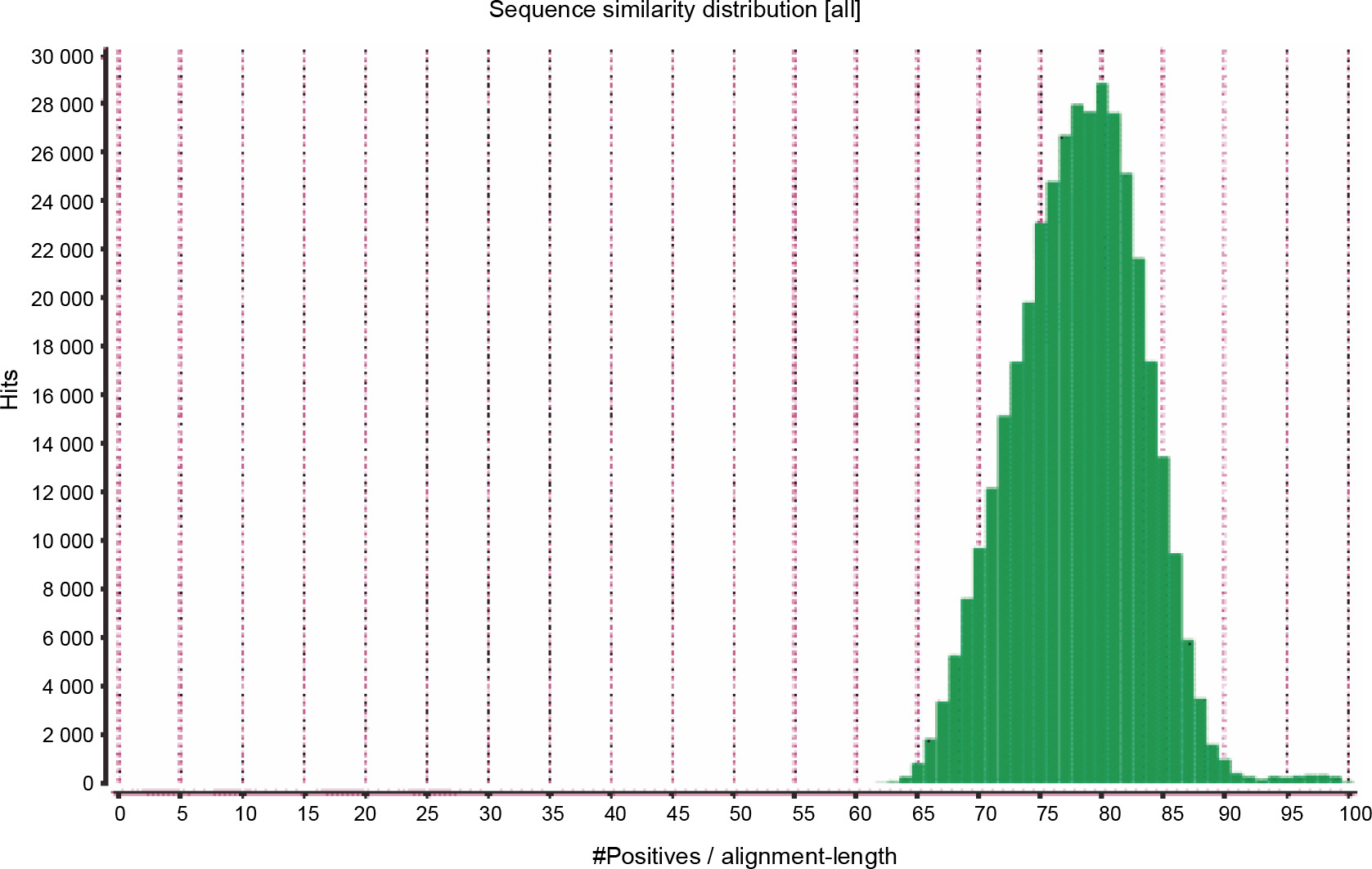
The primary aim of generating a transcriptome assembly for a non-model plant is integrating the transcriptome assembly with functional annotation to reveal its attributes and understand its characteristics. In the present study, to predict the function of the assembled transcripts, similarity comparisons were performed at two levels – sequence-based and domain-based alignments. As mentioned previously, the transcriptome containing 42 528 transcripts outputted by TransDecoder was used for the annotation purpose. All 42 528 transcripts were aligned against the NCBI NR (non-redundant) database. Of them, 91.28% showed significant blast hits (Suppl. file 3 – https://data.mendeley.com/datasets/bxcx9jhzdt). The e-value of 10E-10 was chosen to ameliorate the similarity search output for all blast-based homology annotations. Although an e-value cutoff of 10E-10 was applied for homology search analysis, the e-value for the major fraction of the matched sequences was below 10E-10, as shown in Figure 2, indicating good coherent similarity between the query and the subject. The similarity between the query and the subject began from 65% with a few hits and maximized to 80% with around 30 000 hits (Fig. 3). These distribution values indicated the robustness and accuracy of the assembly.
Fig. 2
The e-value distribution for the blast hits of comparison between the transcripts of Peganum harmala transcriptome and the NCBI NR database; it is an indicator for good coherent similarity between the query and the subject; a low e-value shows better match between the query and the subject
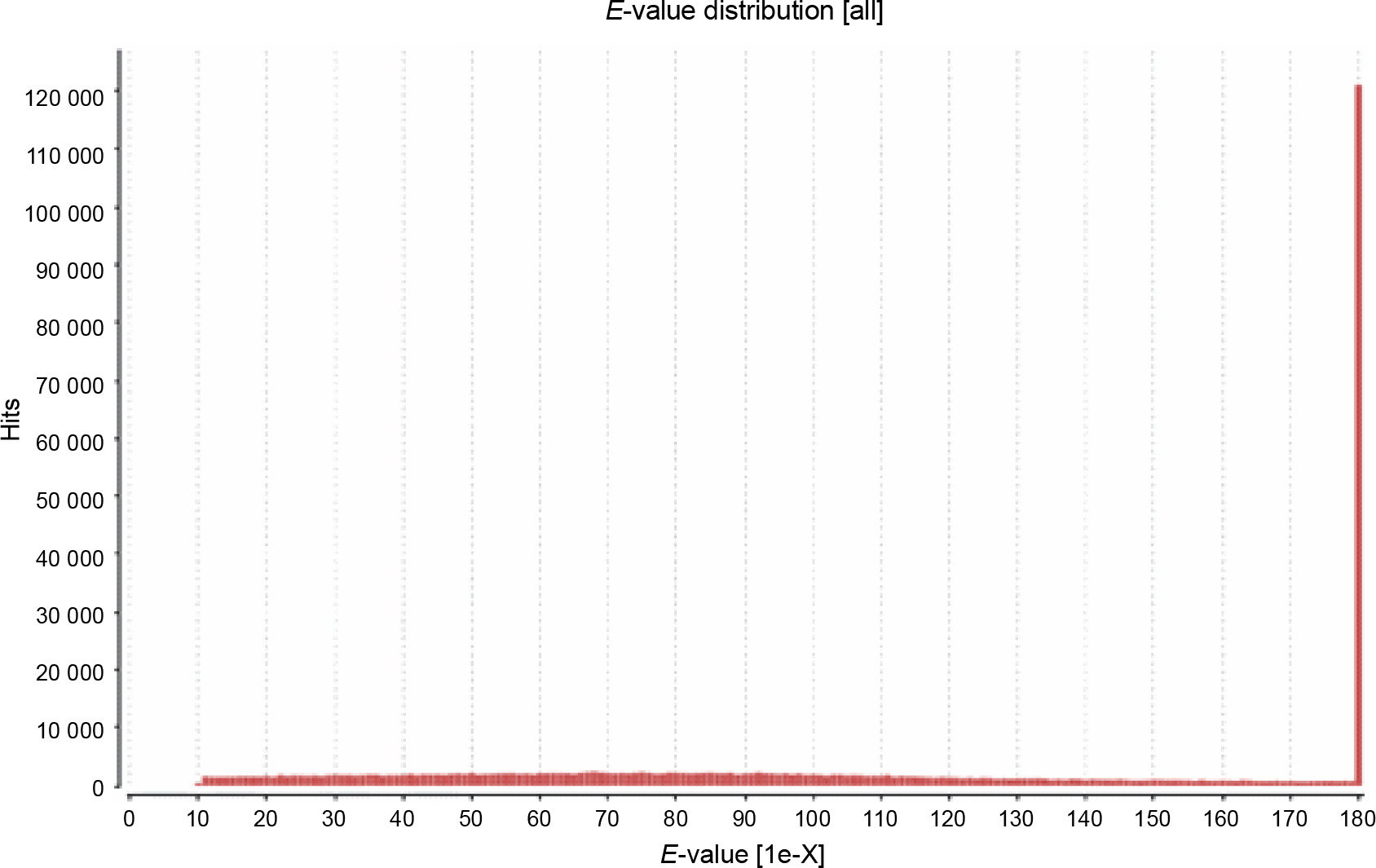
Fig. 3
Similarity distribution among blast hits between the transcriptome of Peganum harmala and the NCBI NR database; the higher number of hits for more positive/alignment length indicates that the query and the subject are more similar, and hence, the generated transcriptome was well structured
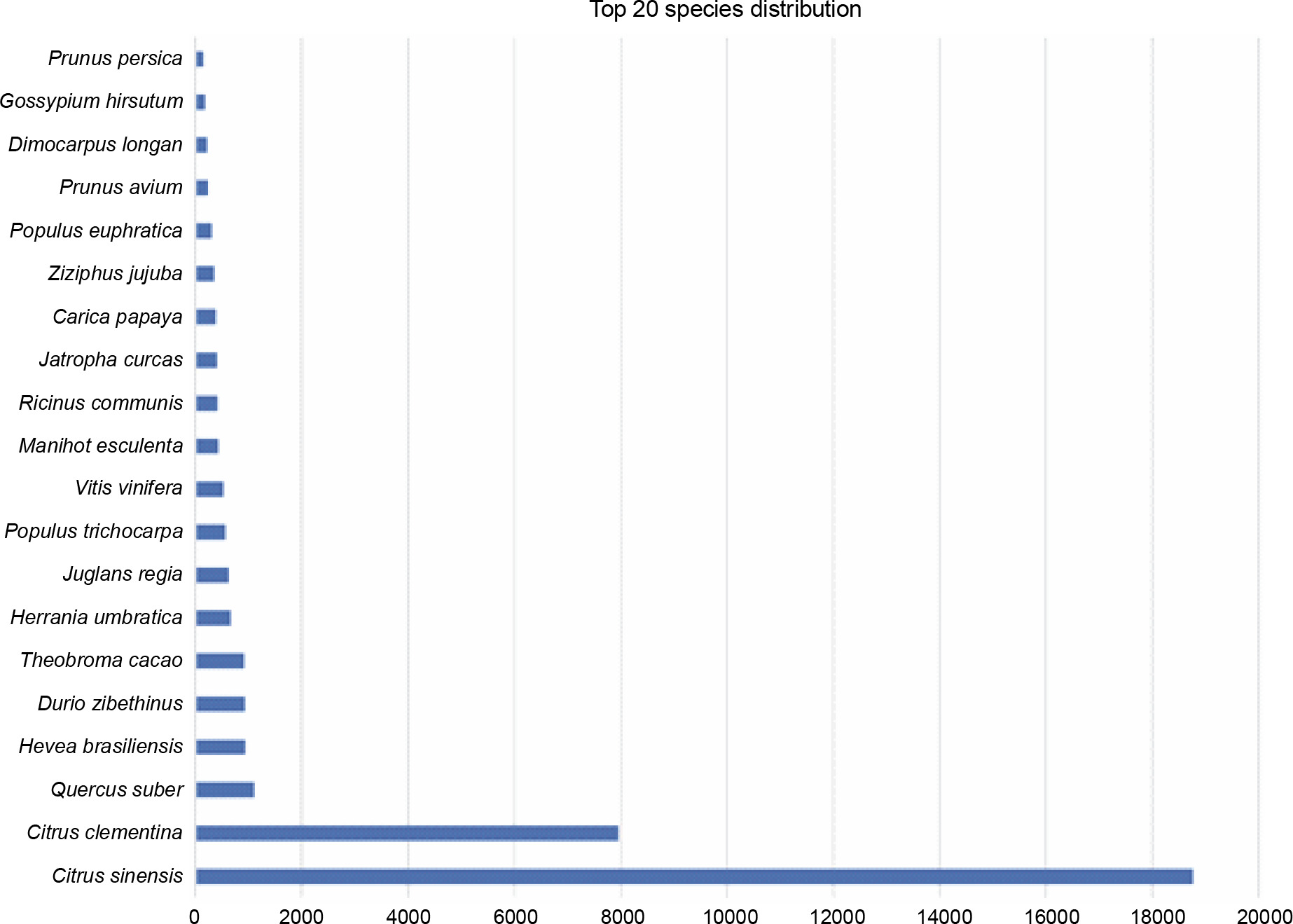
The first best hits for each transcript were listed based on plant species. A total of 3721 hits were without any match, which constituted 8.74% of all transcripts. In total, the transcripts of P. harmala matched with the transcripts of 260 species. Based on the species distribution with the matched transcripts, the highest homology was observed between P. harmala transcripts and Citrus species, with 18 766 hits for C. sinensis (44.12%) and 7952 hits for C. clementina (18.69%), followed by Quercus suber, Hevea brasiliensis, Durio zibethinus, Theobroma cacao, Herrania umbratica, Juglans regia, and Populus trichocarpa (Fig. 4). Interestingly, all species that showed high homology were trees with a natural distribution in temperate and tropical zones. Quercus suber is a member of the Fagales order, whereas H. brasiliensis, D. zibethinus, T. cacao, and H. umbratica are tropical trees of the Malvales order, which is very close to Sapindales (Kubitzki, 2011). Surprisingly, H. umbratica, or Colombian cacao, a species phylogenetically close to T. cacao, also showed high homology (Sousa Silva and Figueira, 2005). It is endemic to the tropical zones of Colombia, whereas P. harmala is found in regions faced with drought, cold, and salinity, but it is not present in Latin America. This phylogenetic closeness is an interesting topic to find biodiversity and evolutionary events based on plant geographical distribution. These results also indicated that Citrus and Pistacia were the closest species to P. harmala as they belong to the same order, Sapindales. Similar species closeness was reported for Citrus and Pistacia vera (Moazzam Jazi et al., 2017). However, as depicted in Figure 4, the species following Citrus (as the species with the most blast hit matches with the transcriptome of P. harmala ) showed less than 2000 hits. If the blast matches with the less aligned bases between the transcripts of P. harmala and those of Citrus are considered, i.e., less identification percentage of resemblance, then more similar transcripts exist between Citrus species and P. harmala. This confirms the species adjacency between P. harmala and Citrus sp. and P. vera.
Fig. 4
Species distribution of blast-based comparison between the transcripts of Peganum harmala transcriptome and the NCBI NT database; the top species belong to Citrus, which is very close to Peganum and Pistacia of the same order
To improve the annotation and gain gene descriptions and further information, the integrated transcriptome of P. harmala was blasted against the SwissProt and UniProt databases. The number of matched proteins in the UniProt database was 38,935, as revealed by the blast against the NR database. As the SwissProt database contains expert-reviewed proteins and has a better resolution and accuracy, the transcriptome of P. harmala was compared with the protein sequences of the SwissProt database using blastx. This comparison showed a total of 29 831 (70% of the transcripts of P. harmala transcriptome) blast hits matched to SwissProt protein sequences with a significant e-value. The most abundant proteins found as UniProtKB hits with more than 20 top-matched hits were Q9SZL8, Q93YU8, Q9ZT94, A0A1P8AUY4, P0C2F6, Q6NQJ8, Q9S7I6, and O23372, as presented in Table 2. These proteins were chosen because they were the most abundant in the generated integrated transcriptome. These proteins are mainly related to gene ontology terms of the ribosome, DNA integration process and the regulation of gene expression during growth, development, and stress responses. In the GO term of DNA integration process, a DNA segment is incorporated into another, usually larger, DNA molecule such as a chromosome. FAR1-RELATED SEQUENCE 5 ((FAR1: FAR-RED IMPAIRED RESPONSE 1; FRS5, Q9SZL8) belongs to subgroup IV of the FRS family and possesses the WRKY-GCM (glial cells missing) zinc-finger DNA binding domain and nuclear localization signal (NLS) in the middle of the N-terminal (Ma and Li, 2018; Lin and Wang, 2004). It is a putative transcription activator regulating the light control of development and functions in many biological processes (BP) such as growth and development and stress resistance (Ma and Li, 2018). Q93YU8 or nitrate regulatory gene 2 protein is involved in nitrate signaling and regulation (Xu et al., 2016). It causes nitrate accumulation in plants by modulating nitrate uptake by roots and nitrate translocation. A previous study showed that nitrate accumulation and hormonal signaling can function toward stress responses in plants (Zhao et al., 2018). As an upstream key gene, it regulates the expression of NIN LIKE PROTEIN 7 and nitrate transporter 1.1, which are involved in nitrogen assimilation (Yu et al., 2016). The findings of the present study based on protein annotation suggest that the high number of genes of the abovementioned proteins that are involved in nitrogen metabolism and regulatory pathways can justify the characteristic of producing alkaloids as nitrogen-based compounds, and these genes can be the key genes in stress mechanisms and alkaloid biosynthesis in P. harmala. These findings also imply that P. harmala is a bioindicator for nitrogen-rich soils. However, further functional genomic studies need to reveal the exact functions of these proteins in P. harmala.
Table 2
The top 20 proteins of Peganum harmala with the highest number of assigned SwissProt protein matches; they were revealed by blastx search against the SwissProt database; Arabidopsis gene IDs were added to precise and complete SwissProt protein name
EggNOG can rapidly generate KO and protein annotation based on KEGG and UniProt as the core data bases (Huerta-Cepas et al., 2019). It revealed 3501 KEGG entries for 14 001 transcripts. The most abundant KEGG entries were K09422 (myb proto-oncogene protein, plant: MYBP), K14638 (solute carrier family 15 (peptide/histidine transporter)), K17964 (leucine-rich pentatricopeptide repeat (PPR) motif-containing protein), K08869 (aarF domain-containing kinase), K15032 (Mitochondrial transcription termination tactor (mTERF) domain-containing protein), K17710 (PPR domain-containing protein 1), and K00430 (peroxidase). These genes are primarily involved in the regulatory system and signaling (Zhang et al., 2019; Gocal et al., 2001; Daniel and Kottra, 2004; Wisidagama et al., 2019; Cui et al., 2019).
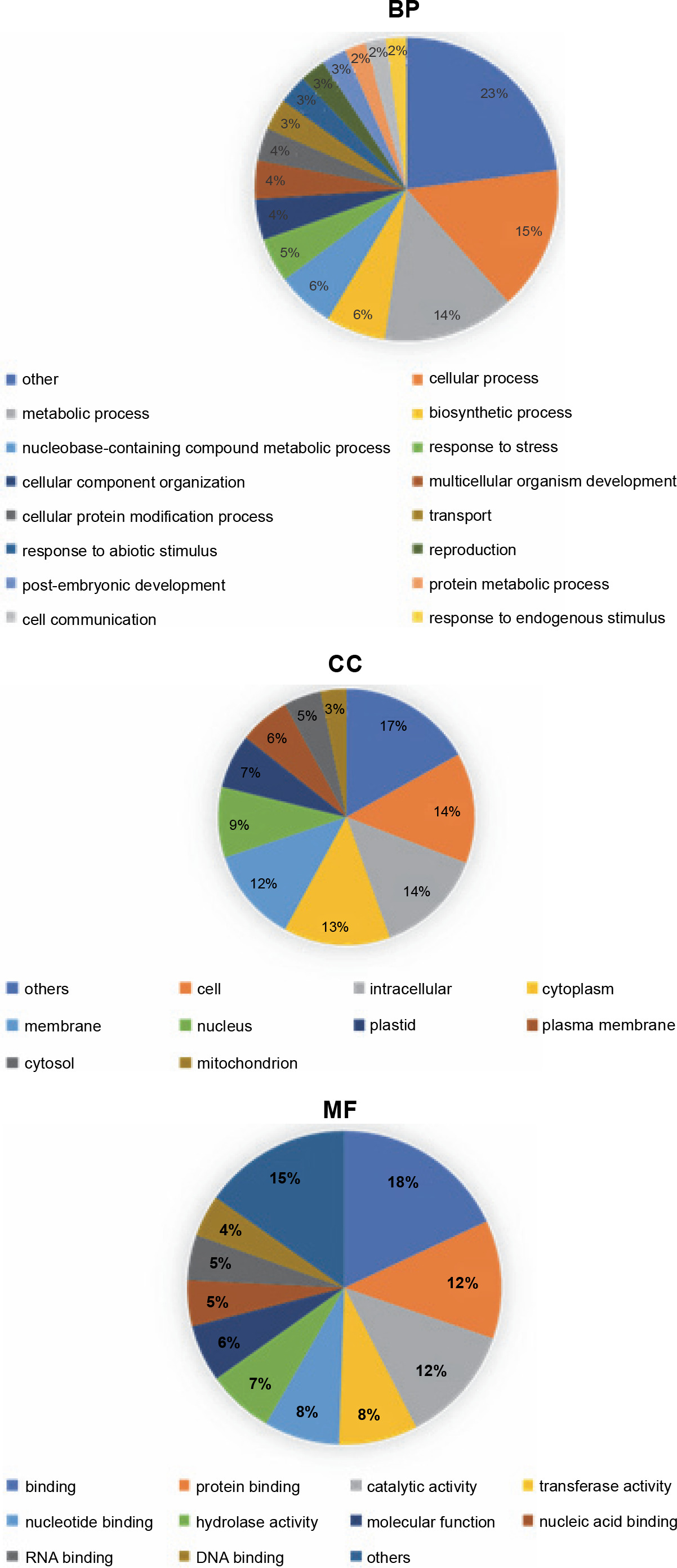
GOanna is an AgBase program used to disclose GO terms by sequence similarity (McCarthy et al., 2006; McCarthy et al., 2007). Among the GO terms for the P. harmala transcripts revealed by GOanna, 33 466 transcripts matched genes and their associated GO terms in the plant database of AgBase. GOslimviewer of AgBase classified GO terms into three categories, namely molecular function (MF), biological process (BP), and cellular component (CC). GO terms with the most hits were binding, protein binding and catalytic activity for MF, cellular process, metabolic process and biosynthesis process for BP and cells, and intracellular and cytoplasm for CC. Figure 5 demonstrates GO categories with more than 3000 annotated transcripts.
Fig. 5
Top GO terms with more than 3000 transcripts and their distribution among all annotated transcripts of the Peganum harmala transcriptome against the Plants database of the GO slimviewer tool; for the BP group, cellular process, metabolic process, and biosynthetic process were the top GO terms; for the CC group, cell, intracellular, and cytoplasm were the top GO terms; for the MF group, binding, protein binding, and catalytic activity were the top GO terms; BP – biological process; CC – cellular component; MF – molecular function
Comparisons between different plant species and P. harmala
To demonstrate the biodiversity relation of P. harmala with different species, the transcriptome of P. harmala was analyzed using blastx comparisons with other species (Table 3). Three species of Citrus showed around 90% of similarity to the transcripts of P. harmala, with C. clementina showing the highest similarity of 90.3% homology. This was expected as these species belong to the Sapindales order. Another species of the Sapindales order, Papaya carica, showed 86.4% similarity with P. harmala. Interestingly, Catharanthus roseus, Eschscholzia californica, Nicotiana tabacum, and Papaver somniferum, which belong to the Brassicales order, showed a close similarity of around 84–85% with P. harmala (Table 3). These alkaloid plants might share a higher similarity with P. harmala because they contain similar nitrogen-based metabolites (Thawabteh et al., 2019; Hussain et al., 2018). However, as these species produce different alkaloid compounds, the similarity between their transcriptomes led us to focus only on the genes involved in alkaloid metabolism and biosynthesis to find any likely relationship between them.
Table 3
Functional annotation summary of Peganum harmala transcriptome; the transcripts of transcriptome of P. harmala were compared with different species by blastx to evaluate the robustness of the generated transcriptome; the selected plant species were phylogenetically close species to P. harmala and alkaloid plants
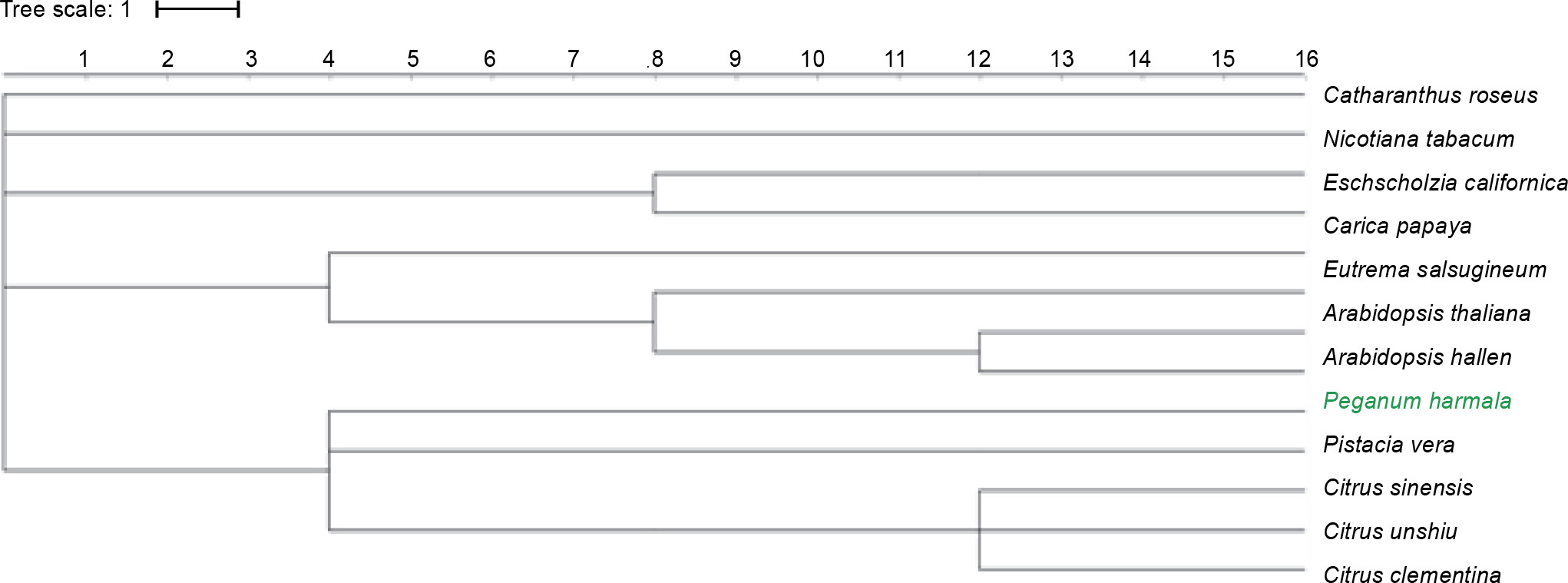
In the comparison of the transcriptome of P. harmala with the reference genome data of A. thaliana (Araport11), 36 377 (85.5%) sequences displayed significant homology. A comparison of the transcripts of P. harmala with A. halleri, a heavy-metal resistant species, revealed 82.8% similarity. These comparisons can help study in detail P. harmala as a model plant. Genealogical trees depict the relationship between species and reveal the phylogenetic closeness between them. The transcriptomic findings of this study are in accordance with the generated genealogical tree (Fig. 6), where these species show a close relationship, suggesting that the P. harmala transcriptome generated in the present study was properly assembled and annotated. The distance in the genealogical tree between P. harmala and P. vera might be due to the lack of publicly available well-documented sequence information for these species.
Fig. 6
The phylogenetic tree for species of Sapindales, Malvales, and Brassicale; Pistacia vera is the closest species to Peganum harmala by 1 tree scale distance, followed by Citrus species; alkaloid plants including Papaver somniferum, Catharanthus roseus, Nicotiana tabacum, and Eschscholzia californica are not close species to P. harmala ; Arabidopsis species are closer to P. harmala than alkaloid-producing plants
On the other hand, the transcripts of P. harmala were compared with different model species that possess alkaloid synthesis ability and belong to the same plant order (Fig. 7). E. salsugineum is a halophyte and is established as a model for studying stress responses because of its likely innate resistant characteristic (Yang et al., 2013). Comparison between the transcripts of P. harmala and E. salsugineum showed a higher similarity than comparing the transcripts of P. harmala with the transcripts of two Arabidopsis species, namely A. thaliana and A. halleri. The transcripts involved in abiotic and biotic stress responses were common specifically between P. harmala and E. salsugineum, whereas for the Arabidopsis species, the common matched transcripts belonged to chloroplast and mitochondrion and were uncharacterized genes (Fig. 7).
Fig. 7
Comparisons between the transcripts of Peganum harmala and the data of other species and datasets revealed by blast; the transcripts of P. harmala and close species of the Sapindales order (A), model plants (B), alkaloid plants (C), and SwissProt, NR and Arabidopsis (D) were compared; the number of transcripts with significant hits is presented in each intersection of the Venn diagram; the most common transcripts were observed between the transcripts of P. harmala and the species of Sapindales
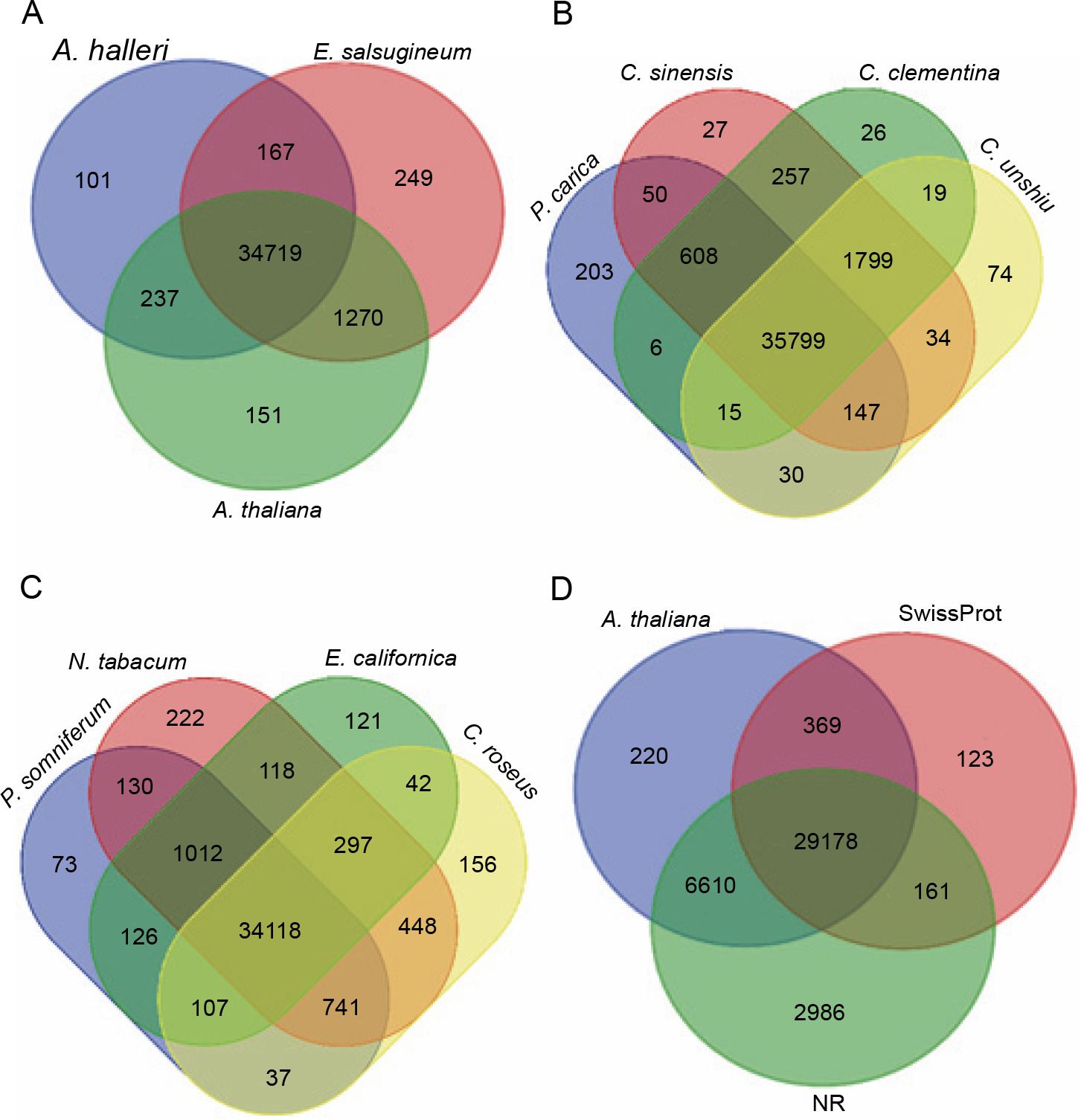
Interestingly, shared transcripts between P. harmala and E. salsugineum possessed various copies (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). The following are a few examples of common genes between P. harmala and E. salsugineum with the highest transcript copy number. ERF061 protein functions as a transcriptional activator modulating gene expression in stress signal transduction pathways (Dubois et al., 2013). ATG8-interacting protein 2 (Autophagy-related protein 8: ATG8) is involved in the vesicle-to-vacuole trafficking pathway induced by salinity and is overexpressed in germinating seeds of Arabidopsis wild-type plants (Michaeli et al., 2014) The functions of “ATG8-interacting protein 2” in germination and stress response make it an interesting candidate gene of P. harmala for further studies. R genes including disease resistance RECOGNITION OF PERONOSPORA PARASITICA RPP13-like, mitogen-activated protein kinase kinase kinase 1-like (MAPKKK1) (Kong et al., 2012) and disease resistance protein Rho-type GTPase-activating 3 (RGA3) and zinc finger A20 and AN1 domain-containing stressassociated protein 5-like are involved in responses to drought and cold (Mukhopadhyay et al., 2004; Sekhwal et al., 2015). Disease resistance in Arabidopsis, wheat, and barley due to the abovementioned proteins has been reported previously (Sekhwal et al., 2015). The high transcript copy number of the abovementioned genes of the transcriptome of P. harmala (Suppl. file 3 – https://data.mendeley.com/datasets/bxcx9jhzdt) suggests that P. harmala possesses an inherent stress-responsive system. However, a profound comparative study between P. harmala and E. salsugineum and further functional genomic analyses are needed to reveal how they function in relation to tolerance to environmental adverse cues.
Among close species belonging to the Sapindales order (Fig. 6) and based on orthologs, Citrus is closer to P. harmala than to Papaya carica, whereas there were more shared sequences (as revealed by blastx) with P. carica than with Citrus species (Fig. 7). Different alkaloid plants, including Papaver somniferum, Nicotiana tabacum, Eschscholzia californica, and Catharanthus roseus, were analyzed using blastx to evaluate the similarity between the transcripts of P. harmala and their genomic data. The results of blastx between these alkaloid plants displayed that N. tabacum shared a higher number of species-specific sequences with P. harmala than any other alkaloid-producing plants (Fig. 7). This higher number of species-specific sequences between P. harmala and N. tabacum (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt) suggests that they might be closer in alkaloid compositions and biosynthesis ability. However, after manually curating, the common transcripts were related to different BPs, MFs, and nonannotated and uncharacterized ones. Therefore, these results need to be confirmed by further functional studies and deepening annotation to disclose similarities between N. tabacum and P. harmala.
The abovementioned findings of a high copy number of stress-responsive transcripts and similarity between P. harmala and other alkaloid plants imply that P. harmala can be a potential stress model plant and also a model plant for alkaloid biosynthesis. These findings suggest that comparative studies between many species sharing a special trait, characteristic, metabolite, or sequence can reveal more precise details on various closely related plants as models. This has been reported for Chlamydomonas reinhardtii (Alfred and Baldwin, 2015), a chlorophyte used as a model due to its photosynthetic genes and ability. Another example is Marchantia, which is used as a model due to its intrinsic ability to absorb heavy metals (Poveda, 2020; Li et al., 2020). Wild plants can be used as models in plant breeding and screening programs as the genome of wild species (not domesticated plants, whose genome is not intact generally) is intact and has not been yet modified and manipulated (Chang et al., 2016). Therefore, the unknown candidate genes for a specific trait of interest can be identified by studying wild plants with nonmodified genome and comparing them with other species. Such a comparison between wild species and other plants (with documented genomic data) allows examining many genes involved in a desired trait.
TFs and regulators
To enrich the transcriptome analysis, regulatory elements and TFs were studied using PlantTFcat, a tool that classifies the sequences based on regulatory and TF families. PlantTFcat categorized 3887 transcripts of the transcriptome of P. harmala as regulatory and transcriptional elements (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). These regulatory transcripts comprised around 9% of the total transcripts of the transcriptome of P. harmala. This is in accordance with previous reports in which generally 5–10% of plant genomic data are TFs and regulatory elements (Mitsuda and Ohme-Takagi, 2009; Jin et al., 2017). However, the number of TFs and regulatory elements of the transcriptome of P. harmala exceeded the reported TFs and regulatory elements of C. sinensis and C. clementina. The transcriptomes of these latter Citrus species contain around 2000 TFs covering approximately 5% of their transcripts (http://planttfdb.gao-lab.org/).
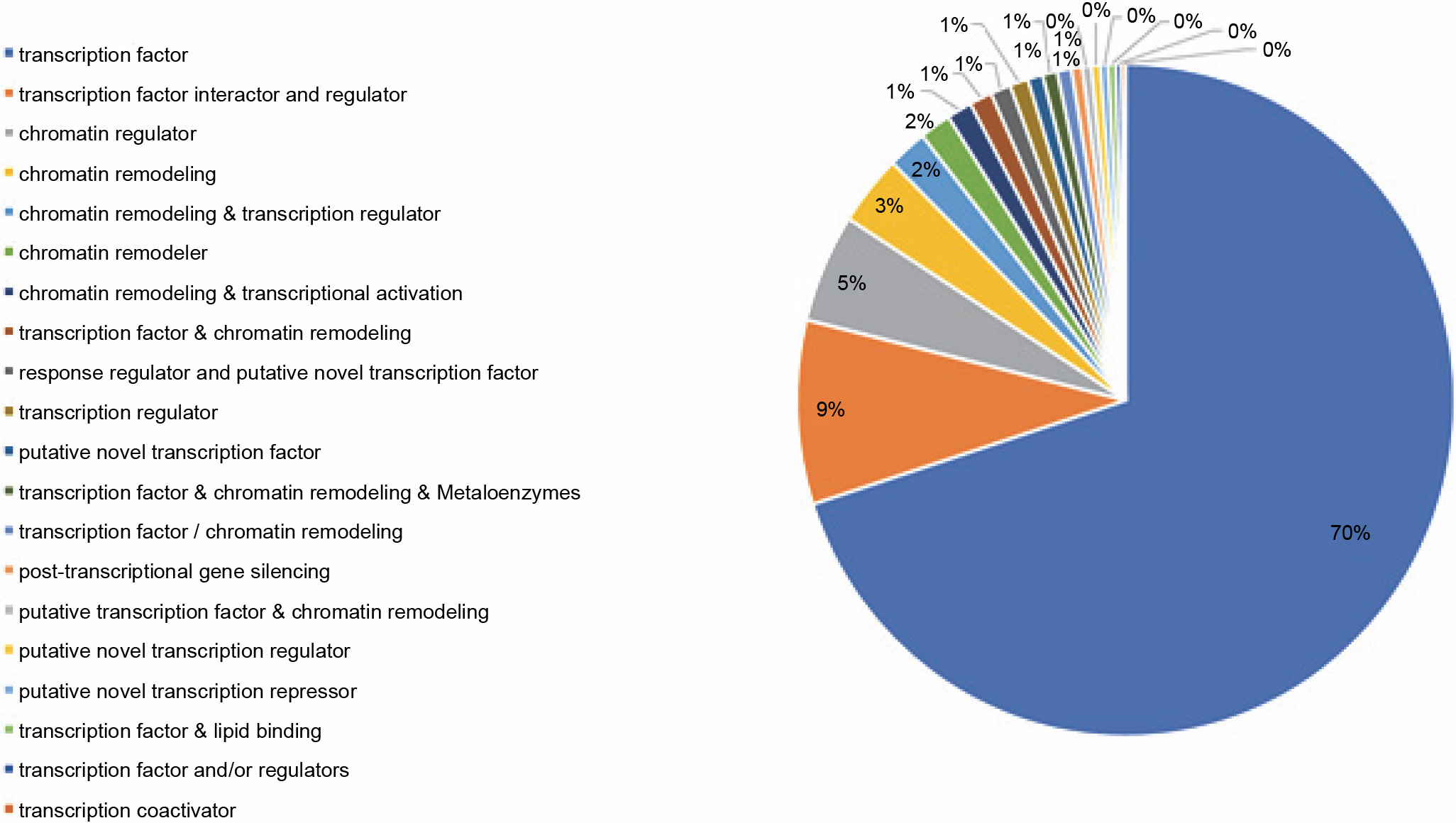
The PlantTFcat tool groups transcription and regulatory elements into different types. The transcriptome of P. harmala was subjected to PlantTFcat analysis, in which 20 PlantTFcat group types within 2721 TF members, 344 of TF interactors and regulators and 203 of chromatin regulators, were observed. These 20 PlantTFcat types were the most abundant (Fig. 8; Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). The results of the PlantTFcat tool also showed 96 TF families, among which C2H2 Zip Finger, WD40-like (W and D stand for amino acids tryptophan-aspartic acid respectively), MYB-HB-like (myeloblastosis-homeobox), PHD (plant homeodomain), C3H, CCHC(Zn) (Cys2HisCys), AP2-EREBP (APETALA2 – ethylene-responsive element binding proteins), and bHLH were the most abundant (Table 4; Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). Interestingly, the most abundant TF families were those whose functions were related to plant stress responses and tolerance enhancement.
Table 4
TF families of the Peganum harmala transcriptome with more than 100 members; these TF families were identified by PlantTFcat
| Family | Family type | Number |
|---|---|---|
| C2H2 | transcription factor (TF) | 584 |
| WD40-like | TF | 443 |
| MYB-HB-like | TF | 214 |
| PHD | chromatin regulator | 203 |
| C3H | TF | 141 |
| CCHC(Zn) | TF interactor and regulator | 140 |
| AP2-EREBP | TF | 114 |
| bHLH | TF | 110 |
Fig. 8
Types of transcriptional elements and regulatory factors of the transcripts of Peganum harmala transcriptome revealed by PlantTFcat; the top types were transcription factor (TF), TF interactor and regulator, chromatin regulator, and chromatin remodeling
A total of 584 members of the C2H2 family are found in the transcriptome of P. harmala. In A. thaliana, 176 C2H2-ZFPs (zin finger protein) have been reported, and 189, 109, 321, 118, and 47 C2H2-type ZFPs have been identified in rice (Oryza sativa ), poplar (P. trichocarpa ), soybean (Glycine max ), tobacco (N. tabacum ), and wheat (Triticum aestivum ), respectively (Liu et al., 2022). Brassica napus and Gossypium hirsutum have 384 and 318 C2H2 members, respectively (Jin et al., 2017). The C2H2 family comprises 0.7% of the Arabidopsis transcriptome (Englbrecht et al., 2004). The results of the present study indicated that 1.4% of transcripts of the transcriptome of P. harmala belonged to the C2H2 family, which is twice higher compared with Arabidopsis data and more than that of all reported plants. This suggests that P. harmala possesses a potential regulatory network based on C2H2 ZFP TFs that can help it tolerate adverse conditions. However, it should be taken into account that PlantTFcat was used to perform TF enrichment annotation as it covers more TF families and contains other regulatory factors that other databases do not have. Furthermore, PlantTFcat uses different algorithms and definitions to determine TF domains and motifs using an ameliorated approach (Dai et al., 2013). The available studies are generally based on other plant TF databases such as PlantTFDB, PlnTFDB, and iTAK.
Some C2H2-type zinc finger proteins are involved in the activation or inhibition of other stress-related genes that enhance tolerance to various stresses. These proteins contain a proline-rich region between the nuclear localization sequence (NLS) and leucine L-box motifs and also the ethylene-responsive-element (EAR) motif (ethylene-responsive element-binding factor associated with the amphiphilic repression domain). These motifs present activation/repression activity to C2H2 proteins (Liu et al., 2022). C2H2 proteins interact with other different zinc finger TFs during stresses to regulate transcription (Kiełbowicz-Matuk, 2012). C-terminus of SALT TOLERANCE ZINC FINGER (STZ), a C2H2 zinc finger protein, functions in the transcriptional activation of dehydration element-binding1A (DREB1A) under salt stress, whereas the DLN-box determines the transcriptional inhibitory activity of C2H2 zinc finger proteins (Han et al., 2020). C2H2 ZF proteins are involved in three stress-signaling pathways, namely abscisic acid ABA-dependent, ABA-independent, and MAPK pathways. In the ABA-dependent pathway, cold-regulated (COR), pyrroline 5 carboxylate synthase, proline transporter, and DREB2A genes are regulated. In the ABA-independent pathway, C2H2 genes modulate peroxidase 24 precursor, plasma membrane receptor-like kinase leaf panicle 2, DREB/CB (C-repeat binding factors), and APx1/2 (ascorbate peroxidase). In the MAPK pathway, C2H2 genes regulate GSH1/2 (glutathione1/2), phytochelating1/2, and CBF1/3 (Liu et al., 2022).
For the WD40 family, a similar pattern was observed in which the P. harmala transcriptome had a higher WD40 transcript content than other plant species. The WD40 TF family is involved in stress-responsive mechanisms (Dietz et al., 2010) and many BPs such as plant development, cell wall formation, immunity, and signaling during stress (Mishra et al., 2012). The WD motif is also known as the Trp-Asp or WD40 motif, indicating tryptophan-aspartic acid consequence in its structure. Generally, plants encode more than 200 putative WDR-containing proteins, including the WD40 domain (Miller et al., 2015). This domain is a molecular structure that starts with glycine and histidine at one extreme. Then, it is followed by a 40-amino acid stretch that ends to tryptophan-aspartic acid (WD repeat) (Villanueva et al., 2016). The findings of the present study showed that the transcriptome of P. harmala included 443 WD40 family members. The higher number of members of the C2H2 and WD40 families of the transcriptome of P. harmala in comparison with other plant species suggests that it is an inherent attribute of P. harmala that enables it to grow in deserts, kavirs (i.e., dry and alkaline areas), and salinity zones. For other TF families, including MYB, bHLH, bZIP, and AP2-EREBP, these findings are in accordance with those of the previous studies where these TF families have been reported among the most abundant TFs of Arabidopsis (Jazayeri et al., 2018; Pireyre and Burow, 2015). However, further studies in functional genomics and molecular biology are needed to prove the hypothesis that higher amounts of these TF members in P. harmala might constitute its inherent resistance to adverse conditions.
On the other hand, the InterPro (IPR) domains of TF families were analyzed to determine their abundance (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). In total, 146 domains were listed, with IPR001841, IPR001965, IPR011011, IPR009057, IPR017907, IPR017986, IPR019786, IPR001005, IPR019787, and IPR018957 being the ten most abundant ones. These IPR domains belonged to zinc finger RING-type, zinc finger PHD-type, zinc finger FYVE/PHDtype (FYVE: Fab 1 (yeast orthologue of PIKfyve), YOTB, Vac 1 (vesicle transport protein), and EEA1), homeoboxlike domain superfamily, WD40-repeat-containing domain, SANT/Myb (SANT: Swi3, Ada2, N-Cor, and TFIIIB) domain, zinc finger, and C3HC4 RING-type. Zinc finger motifs function as part of DNA-binding and protein–protein interaction domains (Li et al., 2022). They form a large regulatory network that senses and responds to different environmental stimuli. Among zinc finger proteins, the C2H2 type is the major group that functions in regulatory networks (Ciftci-Yilmaz and Mittler, 2008). As mentioned above, the C2H2 family is the most abundant in the transcriptome of P. harmala. These results suggest that networks of regulatory domains of zinc finger motifs in DNA and protein-binding interactions evolved in P. harmala to respond effectively to adverse conditions.
Alkaloid-encoding genes
The search for alkaloid-encoding genes and sequences in the NCBI generated a list of 1007 sequences. The Phytozome database search for alkaloid-related genes yielded a list of 3025 peptide sequences. These two lists were combined, and after removing redundancy, the final list included 2478 protein sequences (Suppl. file 1 – https://data.mendeley.com/datasets/bxcx9jhzdt). Using the alkaloid protein sequence dataset compiled from the public domains NCBI and Phytozome and employing blastx, 3124 transcripts of the P. harmala transcriptome matched at least one hit with alkaloid-related proteins (Suppl. file 5 – https://data.mendeley.com/datasets/bxcx9jhzdt). To reveal their function by GO terms, a list of 1336 transcripts matching 633 annotated genes in the UniProt database used in the AgBase collection was generated by GOanna (Suppl. file 4 and Suppl. file 5 – https://data.mendeley.com/datasets/bxcx9jhzdt). The transcripts with the highest copy numbers were MHK10.21 with 14, PAL1 with 14, UBQ3 with 13, RPL8C with 12, and LAC17 with 11 copies. MHK10.21, a member of the copper amine oxidase protein family (EC: 1.4.3.21), is involved in the isoquinoline alkaloid biosynthesis pathway and converts dopamine to 3,4-dihydroxyphenylacetic acid, the key intermediate for the morphinan alkaloid biosynthetic pathway (Lorenz et al., 1988). It is a candidate gene in alkaloid biosynthesis in Lupinus angustifolius (Plewiński et al., 2019). PAL1 (phenylalanine ammonialyase) (EC: 4.3.1.24; InterPro: IPR001106) functions in defense responses to microbial pathogens, in lignin and alkaloid biosynthesis, and in nitrogen metabolism, and its expression was changed in response to pathogens in Capsicum annuum (Kim and Hwang, 2014). These results suggest that P. harmala might employ such genes to achieve alkaloid biosynthesis in a multipurpose manner as a means of stress adaptation. However, further studies are needed that discuss in detail the exact functions of these genes in response to stress and with alkaloid biosynthesis.
GO terms including the alkaloid biosynthetic process, indole alkaloid biosynthetic process, alkaloid metabolic process, and isoquinoline alkaloid biosynthetic process were analyzed. Twenty-five transcripts of the transcriptome of P. harmala were categorized as alkaloid-related transcripts. For the alkaloid-related transcripts, KEGG map analysis showed their corresponding genes (Table 5) to be involved in pathways including tyrosine metabolism [PATH: ko00350] K01593, phenylpropanoid biosynthesis [PATH: ko00940] K13065, stilbenoid, diarylheptanoid and gingerol biosynthesis [PATH: ko00945] K13065, flavonoid biosynthesis [PATH: ko00941] K13065, indole alkaloid biosynthesis [PATH: ko00901] K01593, isoquinoline alkaloid biosynthesis [PATH: ko00950], betalain biosynthesis [PATH: ko00965], dopaminergic synapse [PATH: ko04728], serotonergic synapse [PATH: ko04726], cocaine addiction [PATH: ko05030], amphetamine addiction [PATH: ko05031], alcoholism [PATH: ko05034], cytochrome P450 [BR: ko00199] K23136, exosome [BR: ko04147] K01593, phenylalanine metabolism [PATH: ko00360], tryptophan metabolism [PATH: ko00380], and monoterpenoid biosynthesis [PATH: ko00902].
Table 5
Genes involved in alkaloid biosynthesis pathways; these genes were revealed by a search among GO terms and KEGG maps related to alkaloid biosynthesis
In addition, five transcripts of the P. harmala transcriptome have been found in human pathways, namely dopaminergic synapse, serotonergic synapse, cocaine addiction, amphetamine addiction, and alcoholism, which are related to the nervous system. They belong to aromatic-Lamino-acid / L-tryptophan decarboxylase (DOPA decarboxylase (DDC), L-tyrosine decarboxylase (TDC), Aromatic L-amino acid decarboxylase (AADC)) [EC: 4.1.1.28, EC: 4.1.1.105], which is involved in the metabolism of tyrosine, tryptophan, and phenylalanine. Deficiency of this enzyme leads to serotonin and catecholamine deficiency, resulting in depression (Homan et al., 2015). This finding helps understand how the activity of DDC in the human neuron system is changed by P. harmala usage as an antidepressant (Sassoui et al., 2015).
Interestingly, 11 transcripts matched benzyl alcohol O-benzoyltransferases (EC: 2.3.1.196 and EC: 2.3.1.232 known as benzoyl-CoA:benzyl alcohol/phenylethanol benzoyltransferase (BEBT) and anthraniloyl-coenzyme A (CoA): methanol acyltransferase (AMAT), respectively). They are involved in the conversion of benzoyl-CoA to benzyl benzoate as follows:
The product of the benzyl benzoate reaction is an insect repellent (Hayes and Laws, 2013). Benzoyl-CoA is converted to benzoyl acid (BA), a building block for plant hormones, cofactors, defense compounds, and insect attractants, and is involved in the shikimate pathway (Widhalm and Dudareva, 2015). The shikimate pathway eventually provides substrates for alkaloid biosynthesis (based on Map 01063-6 of KEGG) where BA is used as a substrate. The number of BEBT and AMAT transcripts suggests that benzyl benzoate is the key product for alkaloid biosynthesis and that BEBT is an important gene in P. harmala metabolite pathways. However, further studies are needed to reveal their functions, mechanisms, and subsequent products.
Cytochrome P450 families
Because of their importance in alkaloid biosynthesis as shown in alkaloidal plants such as P. somniferum and E. californica (Hori et al., 2018), the cytochrome P450 group was studied. A search for cytochrome P450 in Araport11 revealed 244 transcripts belonging to this family. Among the 74 P450 subfamilies of Arabidopsis, 46 were matched in P. harmala, covering 257 cytochrome P450 proteins. The richest subfamilies were CYP82C, CYP71B, CYP714A, CYP76C, and CYP86A, with 29, 25, 14, 11, and 11 members, respectively. The CYP82 family has been reported from E. californica as an enriched species for this cytochrome family which is involved in Benzylisoquinoline Alkaloid (BIA) biosynthesis (Hori et al., 2018). This family is found primarily and specifically in alkaloid-producing cells and tissues. A total of 29 members were found for the CYP82C family in P. harmala, which is more than in E. californica. The CYP71B family is involved in monoterpene indole alkaloid biosynthesis and has been reported from C. roseus (Dang et al., 2018). CYP714A is involved in the inactivation of early gibberellin intermediates. CYP86A functions in the biosynthesis of hydroxylated fatty acids required for cutin biosynthesis, cuticle development, and repression of bacterial type III gene expression and is induced by stresses in Arabidopsis, which has five copies of CYP86A (Duan and Schuler, 2005). The findings of the present study indicate that P. harmala is a cytochrome P450-rich plant with an enhanced potential for their usage.
Stress-related transcripts
Stress-related transcripts were mined by a workflow based on GO terms and data of public domains. They were selected to empower the analysis by two different data sources for stress genes. The GO analysis showed 8049 transcripts categorized under terms related to stress, including response to stress, response to an abiotic stimulus, and response to a biotic stimulus. On the other hand, a blast search against the stress sequences from the public domain, i.e., NCBI, returned 12 654 transcripts. Finally, a total of 4853 hits (11.4% of the transcriptome) that are common in both lists were identified as P. harmala stress-related transcripts (Suppl. file 6 – https://data.mendeley.com/datasets/bxcx9jhzdt). The 4853 transcripts were subjected to further functional analyses using PlantTFcat and GOanna to identify their functions.
All 4853 stress-related transcripts belonging to 1556 genes were annotated using GOanna (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). The 10 most abundant genes were as follows: LRR receptor-like serine/threonine-protein kinase GSO1 (GASSHO1), ubiquitin, ultraviolet-B receptor UVR8, LRR receptor-like serine/threonine-protein kinase, FLAGELLIN-SENSITIVE 2 (FLS2), LRR receptor-like serine/threonine-protein kinase RPK2, LOW protein: PPR containing-like protein, LRR receptor-like serine/threonine-protein kinase EF-TU RECEPTOR (EFR), receptor-like protein kinase 5, probable lactoylglutathione lyase, and chloroplastic receptor protein-tyrosine kinase C-TERMINALLY ENCODED PEPTIDE RECEPTOR 2 (CEPR2). They are primarily involved in the perception and signaling of stress. Interestingly, 1014 transcripts related to salt stress were found in P. harmala, justifying the distribution of P. harmala in saline arid zones (Ababou et al., 2013; Karakas, 2020). The number and expression of salt-responsive genes in halophytes are important factors that permit halophytes to tolerate salinity, as observed in different species such as Mesembryanthemum crystallinum with 15% of expressed genes and Porteresia coarctata with 15,158 genes involved in salinity and submergence tolerance (Mishra and Tanna, 2017; Garg et al., 2014). However, these genes of different species belonged to different metabolic pathways, whereas 1014 transcripts of P. harmala are related directly to salinityresponsive processes that were evidenced by GO terms.
Analysis of stress-related transcripts using PlantTFcat revealed 766 transcripts distributed in 56 TF families and 811 TFs and regulatory elements. The most abundant families were as follows: C2H2 with 133 members, MYB-HB-like with 74, WD40-like with 55, AP2-EREBP with 49, NAM with 38, PHD with 38, bHLH with 30, CCHC(Zn) with 28, sucrose nonfermenting 2 (SNF2) with 28, and WRKY with 27 (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). These TF families have been reported in the stress response of other plants. C2H2 TFs are one of the best-studied types of TFs among plants, and they are elevated under salt stress and other abiotic stresses (Kiełbowicz-Matuk, 2012; Liu et al., 2022). Gene expression analysis of rice landrace Horkuch, which is known to have salt tolerance traits, showed a higher expression of MYB family members (Razzaque et al., 2019). In another report, 77 TF families were found for the salt-resistant Kharchia local wheat variety. Of these 77 TF families, WD40-like represented the most abundant category, comprising of 139 unigenes, followed by 107 C2H2 and 39 unigenes representing MYB-HB-like TFs (Goyal et al., 2016). As previously mentioned, these TF families were more abundant in P. harmala than in other plant species. Accordingly, the high number of stress TFs suggests the inherent capacity for stress adaptation and response in P. harmala.
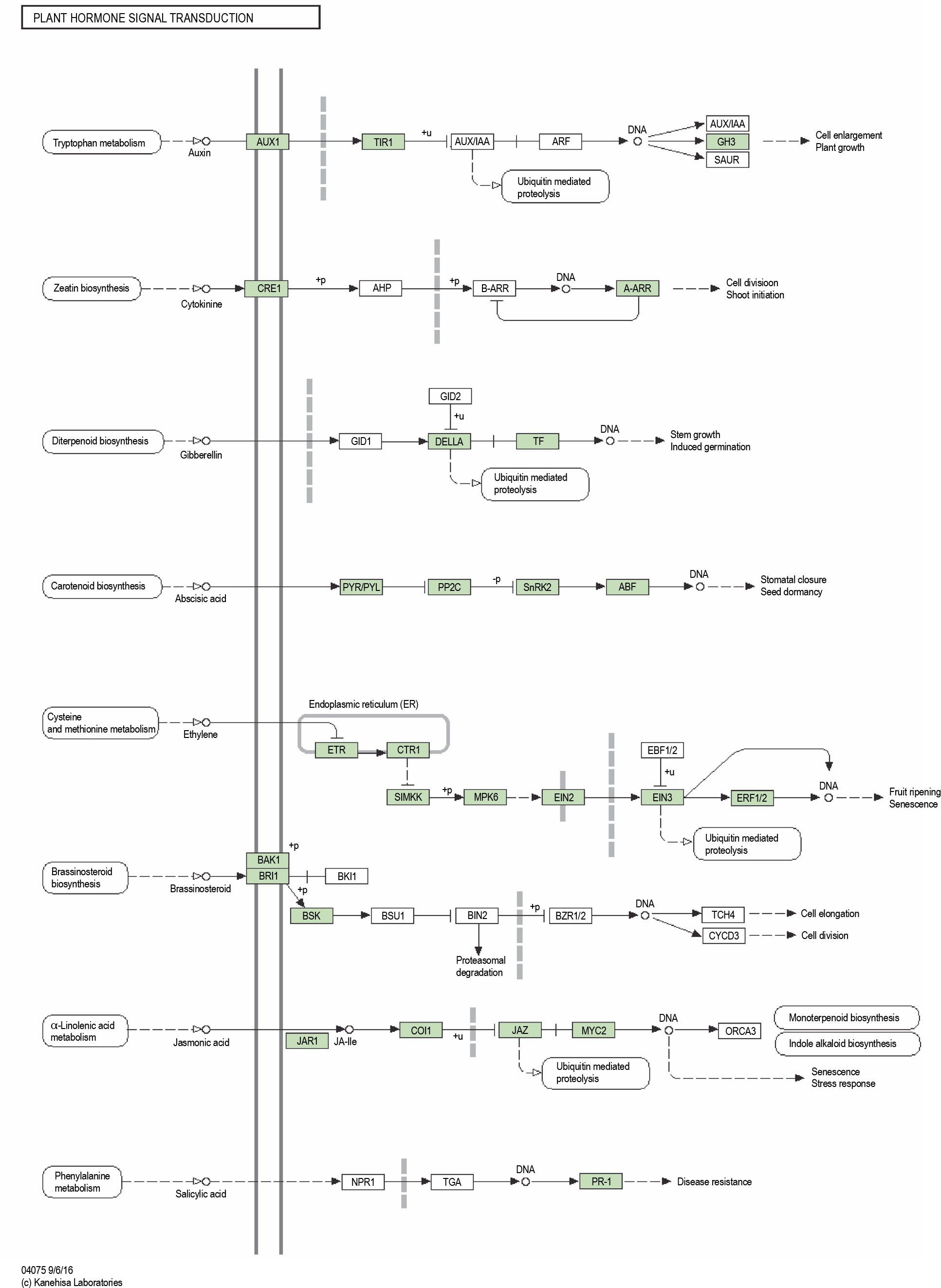
KAAS analysis revealed 3272 KO entries related to stress genes. Two pathways, namely the MAPK signaling with 37 hits and plant hormone signal transduction with 27 members, showed the highest number of transcripts (Fig. 9 and Fig. 10). Out of 78 genes involved in the MAPK signaling pathways, 55 genes were found in the transcriptome of P. harmala. In plant hormone signal transduction, 44 genes were observed, of which 26 genes were found in the transcriptome of P. harmala. MAPK and hormone signaling pathways are closely linked to stress perception and signaling, as shown in Fig. 9 and Fig. 10.
Fig. 9
The MAPK signaling pathway of the KEGG database revealed for the transcripts of Peganum harmala transcriptome; shown it was the KEGG map with the highest number of P. harmala transcript mapped against it; the genes are involved in biotic and abiotic stress responses such as stomatal development, ROS (Reactive Oxygen Species) homeostasis, ethylene biosynthesis, defense response, and adaptation; the green boxes show the gene present in the transcriptome of P. harmala
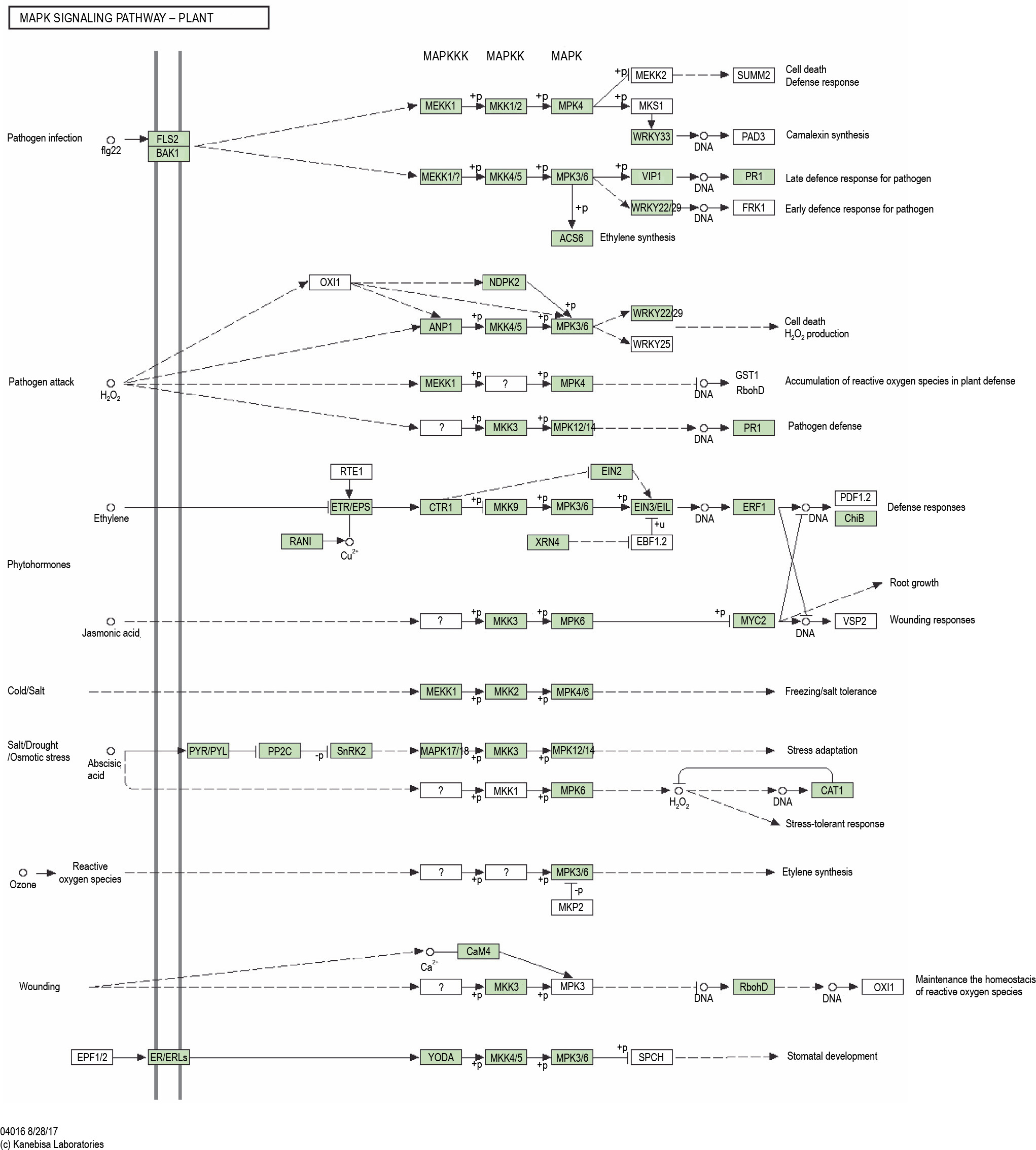
Fig. 10
The plant hormone pathway of the KEGG database revealed for the transcripts of Peganum harmala transcriptome; it was the second pathway with the highest number of P. harmala assigned transcripts; the green boxes show the gene present in the transcriptome of P. harmala
The MAPK pathway triggers a series of threonine/tyrosine and serine/threonine phosphorylation events of other genes. These events are involved in reconfiguring a specific response that is related to transcriptional reprogramming (Taj et al., 2010). The MAPK cascade consists of three parts, namely MAPKKK, MAPKK, and MAPK. MEKK1, MKK1/2, and MPK4 were found in the transcriptome of P. harmala (Fig. 10), which begin three MAPK cascade parts, respectively. Overexpression of these genes of the MAPK signaling pathway and other genes of pathways in other plants has been reported as stress responses, tolerance, and adaptation (He et al., 2020). This can imply that these pathways and their pertinent genes of P. harmala function in favor of its inherent stress tolerance. However, gene network analyses and functional genomic proof are needed to demonstrate the tolerance mechanism in P. harmala via these genes.
In a previous study, 10 genes transcribed into 26 transcripts were revealed as A. halleri species-specific genes involved in heavy metal accumulation and response (Jazayeri et al., 2019). A comparison of the P. harmala transcriptome with A. halleri showed that the transcriptome of P. harmala contains orthologs of the genes of A. halleri. Arabidopsis halleri transcriptome contains two aldehyde dehydrogenase (ALDH) family 2 member transcripts (Araha.11872s0001). The transcriptome of P. harmala showed three transcripts for the ALDH family 2 gene. The ALDH protein family is involved in plant stress responses, and its differential expression has been reported in heavy metal stress (Brocker et al., 2013). The heterologous overexpression of ZmALDH in Arabidopsis increased Al tolerance by promoting the ascorbate-GSH cycle, increasing the transcript levels of antioxidant enzyme genes and the activities of their products, reducing MDA, and increasing free proline synthesis (Du et al., 2022).
Two cysteine synthase 1 genes (Araha.23960s0001) were reported in A. halleri (Jazayeri et al., 2019), and two matching transcripts in the transcriptome of P. harmala were found. Cysteine synthase catalyzes the final step of l-cysteine biosynthesis in plants. Its overexpression leaded to heavy metal tolerance and accumulation in transgenic N. tabacum plants (Kawashima et al., 2004). These orthologs may justify how P. harmala tolerates and responds to heavy metal stress (Ghasemi et al., 2014). They might be the key genes in heavy metal accumulation in P. harmala as it grows in abandoned areas where heavy metals are present in excess (el Berkaoui et al., 2022). As discussed before, the homology search for stress-responsive genes showed a high similarity with E. salsugineum (Fig. 7; Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). These genes are potential candidates for subsequent studies on plant breeding and screening programs and stress mechanism response research. However, further molecular and functional genomic studies are needed to reveal the functions of these genes.
Characterization of SSRs
SSRs are one of the most informative and versatile molecular markers commonly used in genetic diversity evaluation, conservation genomics, and genetic mapping studies (Amiteye, 2021; Jazayeri et al., 2020). Among SSRs, dinucleotides to hexanucleotides were selected because of their more polymorphic character than SSRs of 12–20 bp. Mononucleotide SSRs were not considered as they are prone to errors due to assembly and sequencing. Because of P. harmala ’s ability to tolerate adverse dry and salinity conditions and geographical distribution, identification of SSR markers is highly desirable to identify and conserve its tolerant ecotypes and genotypes; hence, the transcripts of P. harmala were subjected to SSR analysis. At least one SSR was observed in 5.3% of transcripts. For each 1 M bp fragment, ~65 SSRs were found. In total, 2697 SSRs were distributed in 2266 transcripts, whereas 323 transcripts showed more than one SSR (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). Di- and trinucleotide repeats were the most abundant SSRs, accounting for 22.5% and 70.7% of total SSRs, respectively, followed by tetra- (1,3%), penta(0.4%), and hexanucleotide repeats (5.0%). For P. vera, a species closely related to P. harmala, dinucleotide SSRs of transcriptome comprised 44.7% of all SSRs, followed by trinucleotide SSRs with 40.6%, tetranucleotide SSRs with 9.5%, pentanucleotide SSRs with 3.1%, and hexanucleotide SSRs with 2.2% (Moazzam Jazi et al., 2017). Trinucleotide SSRs were the most abundant in P. harmala transcripts, whereas dinucleotide repeats were the most abundant SSRs for P. vera. This indicates that SSRs of close species are not necessarily very similar because of genetic variation between each species. The most abundant SSRs were TC (237), CT (125), GAA (109), TCT (109), AAG (106), AG (104), and CAG (103). With respect to sequence complementarity, the most frequent SSRs were AAG/CTT (554), AG/CT (504), ATC/ATG (318), AGC/CTG (260), ACC/GGT (215), and AGG/CCT (204) (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt).
To reveal stress-related SSRs of genes, the annotation for the transcripts possessing SSRs was performed (Suppl. file 3 – https://data.mendeley.com/datasets/bxcx9jhzdt). Among the genes found were zinc finger A20 and AN1 domain-containing stress-associated protein 3, stress response protein NAC SECONDARY WALL THICKENING PROMOTING FACTOR1 (NST1), heat stress TF A-5-like, abscisic stress-ripening protein 1-like, ERF036, ERF061, ERF104, ERF RAP2-13, ETHYLENE INSENSITIVE 3-like, cold-responsive protein kinase 1-like, heat shock proteins including HSP70, HSP80, and HSP90. These genes are primarily involved in different stress signaling and regulation processes (Giri et al., 2011; Mitsuda et al., 2005; Bourgine and Guihur, 2021; Müller and Munné-Bosch, 2015; Balakireva et al., 2018).
To assure the usefulness of the assigned SSR markers, Primer 3 was applied to design primer pairs for each transcript of SSRs. A total of 2228 primer pairs were generated from the SSR sequences with sufficient flanking sequences and as the best match for each SSR (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). Primer pairs for each SSR sequence were amplified using in silico PCR by FastPCR, confirming their robustness and suggesting the specificity of the corresponding SSR marker. A previous study has reported that SSR markers that generate one in silico PCR product should be putative single-locus markers and could be especially useful (Shi et al., 2014). This large-scale marker discovery facilitates future research for finding stress-tolerance-related markers.
MicroRNA prediction
psRNATarget generated 214 miRNA families with 50 903 microRNA targets for 21 906 transcripts of P. harmala (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). Among them, miR169 (18 matches), miR156 (15), miR166 (10), miR172, miR157, and miR399 showed more than seven matches, followed by miR167, miR171, miR395, miR398, miR5645, and miR8167 with six microRNA each. These results are in accordance with data for Arabidopsis miRNA families, where miR169, miR156, and miR166 were the most abundant miRNA families, with 14, 8, and 7 members, respectively (Szyrajew et al., 2017). Acidic leucine-rich nuclear phosphoprotein 32, defective kernel (DEK-like) protein, pentatricopeptide repeat-containing protein (At4g31850), homeobox-DDT (DNA binding homeobox and Different Transcription factors) domain protein RLT3, and auxin transport protein BIG were the top transcript targets of microRNA (Suppl. file 4 – https://data.mendeley.com/datasets/bxcx9jhzdt). These genes are involved in different BPs such as stress response, growth and development, and regulation. Acidic leucinerich nuclear phosphoprotein 32 is involved in chromatin modification and remodeling, apoptotic caspase modulation, protein phosphatase inhibition, and regulation of intracellular transport in mammalian development (Reilly et al., 2014), but its function has not been yet reported in plants. Protein DEK-like, pentatricopeptide repeat-containing protein, homeobox-DDT domain protein RLT3, and auxin transport protein BIG are involved in transcriptional regulation, and gene expression modulation in growth, development, and stress response (Li et al., 2021; Xing and Xue, 2012). These genes are interesting candidates for further studies in plants. These microRNAs have opened a new avenue for subsequent research on P. harmala to reveal their functions related to different mechanisms and pathways, which are representative attributes of P. harmala.
Conclusions
The present study was conducted to generate the first P. harmala transcriptome. The method used to generate the transcriptome was successful as confirmed by sequence-based parameters, back-mapping sequences, annotation to the public domain, comparative analysis with close species, and GO analyses. Compared with other plant species, a higher copy number of genes involved in the stress response of P. harmala implies that it is a tolerant species per se. It can be used as a model plant for studying environmental stresses, bioindication of soil and geographical zones, and alkaloid biosynthesis. In the case of regulatory elements and TFs, two families, namely C2H2 and WD40-like, showed a high number of differences between P. harmala and other plant species. These two protein families are involved in stress responses, and their exact functions in this species need further functional analyses. Furthermore, as P. harmala grows naturally in noncontrolled areas and abandoned regions and survives under adverse conditions, it can be an adequate source for the search of resistance genes. P. harmala is an alkaloidal plant that produces different nitrogen-based metabolites with wide-ranging applications. Some alkaloid biosynthesis genes of P. harmala such as CYP82C, CYP71B, MHK10.21, PAL1, UBQ3, and RPL8C indicate that it has a great potential for studies on alkaloid production and has medicinal potentiality. With respect to BIA biosynthesis, P. harmala can be used as an alternative for other alkaloid-producing plants. However, this needs further molecular and genomic studies. Other data presented for P. harmala, such as microRNA, SSR, and TFs, provide a good source for subsequent analyses on stress, adaptation, biodiversity, and pharmaceutical screening. The robustness of the generated transcriptome of P. harmala was validated using annotation and comparisons, which indicated that the generated transcriptome can be representative of the species and provides a robust source for further research.