







Introduction
Staphylococcus aureus, a gram-positive opportunistic pathogen, has become a growing health concern because of its ability to cause a myriad of infections in both hospital and community settings and its potential to develop antibiotic resistance at an alarming pace (Lakhundi and Zhang, 2018; Gajdács, 2019; Turner et al., 2019). Researchers worldwide are actively pursuing the search for a protective vaccine against S. aureus, but have failed to develop successful vaccine (Giersing et al., 2016; Parker, 2018; Miller et al., 2020). Many antigens of S. aureus have been tested as vaccine candidates, but none translated into a completely protective vaccine (Fowler and Proctor, 2014; Giersing et al., 2016; Missiakas and Schneewind, 2016; Ansari et al., 2019). Most of the antigens tested to date were targeted to the humoral branch of the immune system that relies on the protective efficacy of antibodies (Karauzum and Datta, 2017; Ansari et al., 2019; O’Brien and McLoughlin, 2019). Despite inducing heightened antibody responses and, in many cases, protection against lethal doses of S. aureus in murine models, these antigens did not prove to be successful in human trials (Fowler et al., 2013; Fattom et al., 2015; Proctor, 2015; Lee et al., 2018).
The lack of clinical success with vaccine candidate antigens is mainly attributed to the incapability of the produced antibodies to induce effector responses that are strong enough to kill the bacteria. Research data suggest that antigens that produce antibody responses alone may not be sufficient to protect from an infection (Fowler and Proctor, 2014; Brown et al., 2015; Yu et al., 2018). This has triggered an interest in antigens that can induce cell-mediated immune responses, especially those mediated by T helper (Th) cells or CD4+ T cells. There is increasing evidence for the protective role of Th cells in human S. aureus infections (Spellberg and Daum, 2012; Lawrence et al., 2012; Brown et al., 2015; Kolata et al., 2015; Yu et al., 2018; Zhang et al., 2018). Memory Th cells were shown to be protective in invasive infections (Brown et al., 2015). CD4+ T cells produce cytokines that enhance the bactericidal activity of neutrophils and macrophages and is critical in resolving a bacterial infection (van Kessel et al., 2014; Brown et al., 2015; Bröker et al., 2016). While it is difficult to correlate the presence of high titer of staphylococcal antibodies to the clinical outcome of an infection (Fowler and Proctor, 2014; Pollitt et al., 2018), impaired cellular immunity has been consistently correlated to the increased risk of developing S. aureus infection (Wiese et al., 2013; Misstear et al., 2014; Yu et al., 2018). Adoptive transfer of activated CD4+ T cells protected mice from lethal dose of S. aureus, but neither B-cells nor antibodies were protective (Joshi et al., 2011).
In the present study, the secreted protein fraction of the staphylococcal proteome was screened for the presence of CD4+ T cell antigens. Secreted proteins have been recognized as vaccination targets in other bacteria and are known to activate CD4+ T cells (Li et al., 2000; Li et al., 2015). It is argued that secreted virulence factors of S. aureus might be better targets for vaccination than cell surface proteins (Spaulding et al., 2013; Salgado-Pabon and Schlievert, 2014). Furthermore, studies in rabbit models have proved that a multivalent vaccine targeting the secreted virulence factors of S. aureus protects against life-threatening infections (Spaulding et al., 2013). Moreover, many secreted proteins of S. aureus, including superantigens, cytolysins, and enterotoxins, are critically involved in the cause and progression of severe illnesses such as necrotizing pneumonia, sepsis, and infective endocarditis (Pragman et al., 2004; Bubeck-Wardenburg et al., 2007; Strandberg et al., 2010; Kong et al., 2016).
CD4+ T cells are activated by endogenously cleaved peptides from proteins presented on the HLA (Human Leukocyte Antigen) class II molecules. HLA class II molecules are loaded with proteins of extracellular origin (Holling et al., 2004; Miller et al., 2015); hence, they are associated with the immune presentation of extracellular pathogens such as S. aureus. The binding affinity of peptides generated from a protein to a HLA class II molecule is a critical determinant of its ability to induce a T cell-mediated immune response. In the present study, a pool of 59 secreted proteins (referred to as “Secretome”) from S. aureus COL I was analyzed computationally to identify peptides that bind to HLA class II molecules by using a combination of dataset-based predictive algorithm and structure-based modeling. A bioinformatics-based screening for CD4+ T cells offers two key advantages. First, as the HLA molecules are highly polymorphic, allele-specific variations in HLA binding have to be considered while identifying vaccine candidates with wide population coverage. This can be achieved using a computational screen as a good number of HLA allelic variants are available in databases. Second, computational screening is performed using human HLA sequences and hence may provide more accurate and clinically translatable hits than animal models with partial similarity to human HLA sequences.
Methods
Prediction of HLA class II binding peptides from the secretome
The secreted proteins from the culture supernatant of S. aureus subsp. aureus COL identified using two-dimensional LC-MS in an earlier study (Ravipaty and Reilly, 2010) were used as the starting dataset for this study. The amino acid sequences of the 59 proteins were retrieved from NCBI GenBank (accession number: CP000046.1). HLA class II binding peptides from the proteins were predicted using the NetMHCII 2.2 prediction server (www.cbs.dtu.dk/services/NetMHCII/) (Nielsen et al., 2007; Nielsen and Lund, 2009). The server predicts the binding of peptides to 14 HLA-DR alleles covering the 9 HLA-DR super types, six HLA-DQ, and six HLA-DP alleles by using artificial neural networks. The protein sequences were fed to the server in FASTA format. All possible 15-mer peptides were generated from each protein. The binding affinities of each peptide to the 26 alleles were predicted. The server assigns a core region consisting of 9 amino acids for each peptide. The binding affinity is measured in terms of IC50 expressed in nanomoles (nM) and as a percentage rank to a set of 1 000 000 random natural peptides. The peptides were differentiated into strong binders, weak binders, and nonbinders according to binding affinity. The binding threshold for the strong binding peptide was 50 nM and that for the weak binding peptide was 500 nM. All peptides that bound with IC50 value higher than 500 nM were considered as nonbinders. Only the strong binding peptides were used for further analysis. The number of strong binding peptides and the number of alleles to which each peptide binds were calculated.
Identification of human self peptides
It is important to identify and eliminate peptides with sequence similarity to human protein motifs from the pool of peptides analyzed for vaccine efficiency. The strong binding peptides were aligned for sequence similarity with the nonredundant protein sequence database of Homo sapiens (taxid: 9606) using the BLASTp algorithm (Altschul et al., 1997). The blast hits were then sorted using a BLAST parser algorithm by setting the gap percentage value to zero. The peptides were ranked based on percentage of similarity to human protein motifs. Peptides that showed more than 50% sequence identity were considered self peptides and excluded from the analysis.
Structure-based modeling of peptide-HLA interactions
Non-self peptides that bound to at least one HLA-II allele with IC50 value smaller than 50 nM were used for structure-based modeling of peptide-HLA interactions. The core regions of all the selected peptides were assigned tertiary structures using the PEPstr server (Kaur et al., 2007). The tertiary structures were predicted by assigning a hydrophilic environment.
Protein and peptide preparation
The atomic coordinates of 13 HLA class II alleles bound to resident peptides in the antigen binding groove were obtained from Protein Data Bank (PDB). Table 1 shows the PDB IDs of the crystal structures used for the study and the HLA loci they represent. Seven of the structures were of HLA DR alleles, one was of HLA DP allele, and five were of HLA DQ alleles. The crystal structures were prepared for docking using the protein preparation wizard of Schrödinger suite. Water molecules at a distance beyond 5 Å from the active site residues were removed from the crystal structures, and polar hydrogens were added. The protein structure was then minimized by applying the OPLS_2005 force field until the root mean square deviation (RMSD) of the minimized structure relative to the crystal structure exceeded 0.30 Å. The peptides modeled using the PEPstr server were prepared for docking using the ligprep module of Schrödinger suite by applying the OPLS_2005 force field.
Molecular docking
A docking grid was generated for each HLA class II protein by using the receptor grid generation option of the glide module of Schrödinger suite. The receptor grid box was prepared by keeping the centroid of the resident peptide as the center of the box. The size of the box was set at the minimum that can accommodate a ligand similar in size to the resident peptide. The resident peptides were excluded from the grid. All the prepared peptides were then docked to each of the HLA class II alleles in a three-step process by using the ligand docking option of the glide module of Schrödinger suite. Initially, a High-Throughput Virtual Screening (HTVS) was performed with all the peptides against each HLA-II allele. The peptide poses for each HLA class II allele with glide scores better than –10 kcal/mol were used for standard precision (SP) docking to that allele. The peptide poses that docked with glide scores better than –10 kcal/mol were then docked using the extra precision (XP) module to the corresponding allele.
Control studies
As a control exercise for the peptide-HLA docking, the resident peptides in the crystal structures were removed and docked back to the corresponding HLA molecule, and the glide scores were compared to those of the predicted peptides. The sequences of the resident peptides were obtained from Protein Data Bank. The core sequence and binding affinity were predicted using the NetMHCII 2.2 prediction server. The core sequences were then modeled using the PEPstr server. The peptides were prepared as explained above and docked to the glide grid of the corresponding HLA class II allele with XP precision.
Results
HLA class II allele binding profiles of the generated peptides
A total of 16 724 peptides were generated from the 59 proteins of the secretome by the NetMHCII server, of which 6991 peptides (41.8%) were predicted to bind to at least one HLA allele with an IC50 value below 50 nM (strong binders). The percentage of strong binding peptides generated by individual proteins varied from 26.7 to 78.9%. There was no correlation between the size of a protein and the number of strong binding peptides generated. A summary of the binding profiles of the peptides generated from the proteins based on the analysis of NetMHCII prediction results is provided in supplementary materials 1.
Identification of self and partial self peptides
It is important to identify self and partially self peptides from the pool of strong binding peptides for using them as vaccine candidates. Self and partially self peptides may trigger autoimmune responses if administered in high doses. All the 6991 peptides that were predicted to bind with an IC50 value of less than 50 nM were aligned to the human proteome to classify them into self, partially self, or non-self peptides. Peptides that showed sequence similarity at 9 amino acid positions (60% similarity) or more were considered self peptides. Peptides showing similarity at 7 or 8 positions (45–60% similarity) were considered partially self peptides. Those peptides showing amino acid similarity at less than 7 positions (less than 45% similarity) were considered non-self peptides and were retained for further analysis. Only 545 of the 6991 peptides were non-self peptides. The percentage of strong binding non-self peptides generated by individual proteins varied from 0 to 11.6%. The strong binding non-self peptides are listed in Supplementary materials 2A.
Specificity and promiscuity in peptide-HLA binding
HLA genes are amongst the highest polymorphic genes present in the human genome. Frequencies of HLA molecules are highly variable across human populations. Hence, the specificity and promiscuity of peptide binding to HLA alleles have to be considered during rational design of a vaccine. The relative frequencies of HLA class II alleles can be correlated to the efficacy of a vaccine candidate in a population (Ndung’u et al., 2005). Peptides showing the highest allele coverage (promiscuous peptides) will show the maximum population coverage when included in a vaccine formulation. On the other hand, the analysis of specificity of peptide binding to HLA class II alleles will help to design vaccine formulations tailored for specific populations. In the present study, among the strong binding non-self peptides, 45.5% (248) bound only to a single allele; they were considered as monoallele-specific. Sixty four peptides bound to 5 or more alleles (Supplementary materials 2A). The highest number of alleles bound by a non-self peptide was 13 of the 26 alleles studied. Another peptide showed specificity to 9 alleles. Nine peptides showed specificity to 8 alleles (refer to Table 2).
Table 2
CD4+ T cell epitope vaccine candidates based on NetMHCII prediction
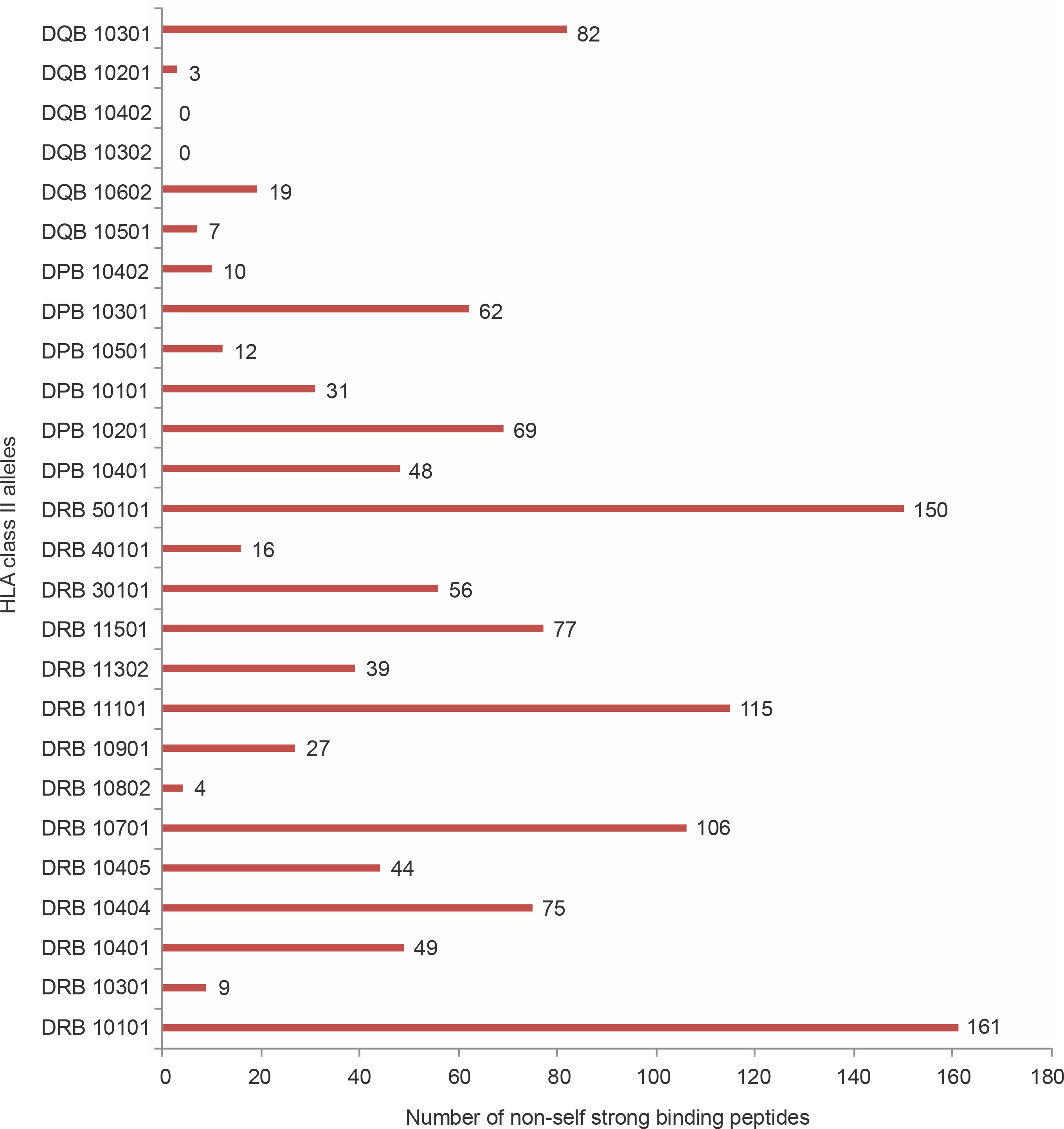
With regard to allele-specific binding, the HLA-DR class of alleles bound the highest number of non-self peptides (66.28 peptides per allele) followed by HLA DP (38.66 peptides per allele) and HLA DQ (18.5 peptides per allele). Although this analysis is arguably influenced by the dissimilar numbers of DR, DQ, and DP alleles used in the study, there were differences in the binding specificities of the peptides within each HLA subclass. HLA DRB1_0101 bound to 161 peptides, i.e., 29.54% of all strong binding non-self peptides. DRB5_0101 bound to 150 peptides (27.5%), while DRB1_1101 bound to 115 peptides (21.1%). Among the HLA DP alleles, DPB1_0201 bound the highest number (69, 12.66%) of peptides followed by DPB10301 (62, 11.37%). Among the HLA DQ alleles, DQB10301 (89, 16.3%) bound the highest number of alleles. HLA DQB10302 and DQB10402 did not bind to non-self peptides. Figure 1 shows the number of peptides bound by each of the allele studied.
Identification of vaccine candidate epitopes
On the basis of the analysis performed on the CD4+ T cell epitopes predicted by the NetMHCII server and the BLAST analysis, non-self strong binding peptides were identified. Among these peptides, a few peptides showed good allele coverage. Peptides with higher allele coverage may be expected to show better population coverage when formulated as a vaccine. Table 2 shows the non-self strong binding peptides with the best allele coverage (≥ 8 alleles). These peptides can be considered as promising CD4+ T cell epitopes that can be used as vaccine candidates. The proteins that generated potential CD4+ T cell epitopes include putative serine-aspartate repeat protein H (SdrH), secretory extracellular matrix and plasma-binding protein (Empbp1), staphylococcal enterotoxin, staphylococcal enterotoxin type B (Seb), and IgG binding protein (Sbi).
Identification of proteins with high epitope density and allele coverage
Proteins that harbor higher number of strong binding non-self peptides may trigger significant CD4+ T cell immune responses in vivo and hence can be considered to act as potential whole protein vaccine candidates. From the analysis of the epitopes generated from each of the source proteins, putative CD4+ T cell protein antigens were identified by calculating the epitope density. Epitope density was calculated as the ratio of the number of non-self strong binding peptides to the total number of peptides generated. Proteins with higher epitope density are more likely to be immunogenic. The highest epitope density was shown by SACOL2557, a conserved domain protein, which produced 15 non-self strong binding peptides from a total of 129 peptides generated. High epitope densities were also exhibited by SACOL0907 (staphylococcal enterotoxin type B, Seb), SACOL0887 (staphylococcal enterotoxin type I, Sei), and SACOL08581 (secretory extracellular matrix and plasma-binding protein, Empbp1). Considering the polymorphic nature of HLA molecules, it is important to identify proteins that produce many epitopes that bind to many different HLA II alleles. The analysis revealed that SACOL0907 (staphylococcal enterotoxin type B, Seb) bound to 16 different HLA alleles; SACOL2019 (SdrH protein, putative (SdrH)) bound to 15 alleles; SACOL2418 bound to 13 alleles; SACOL0442, SACOL1868, and SACOL 08581 bound to 12 alleles; and SACOL1173, SACOL2419, SACOL0887, and SACOL1970 bound to 11 alleles each. The proteins that can evolve as promising vaccine candidates considering both epitope density and allele coverage are listed in Table 3. SACOL 2557 despite showing high epitope density showed poor allelic coverage. On the other hand, many proteins, including SACOL2019 and SACOL2418, showing good allelic coverage had poor epitope density. Eight proteins in the secretome did not produce non-self strong binding peptides.
Table 3
CD4+ T cell protein antigens identified based on epitope density and allele coverage
Molecular feasibility of peptide-HLA class II molecule binding
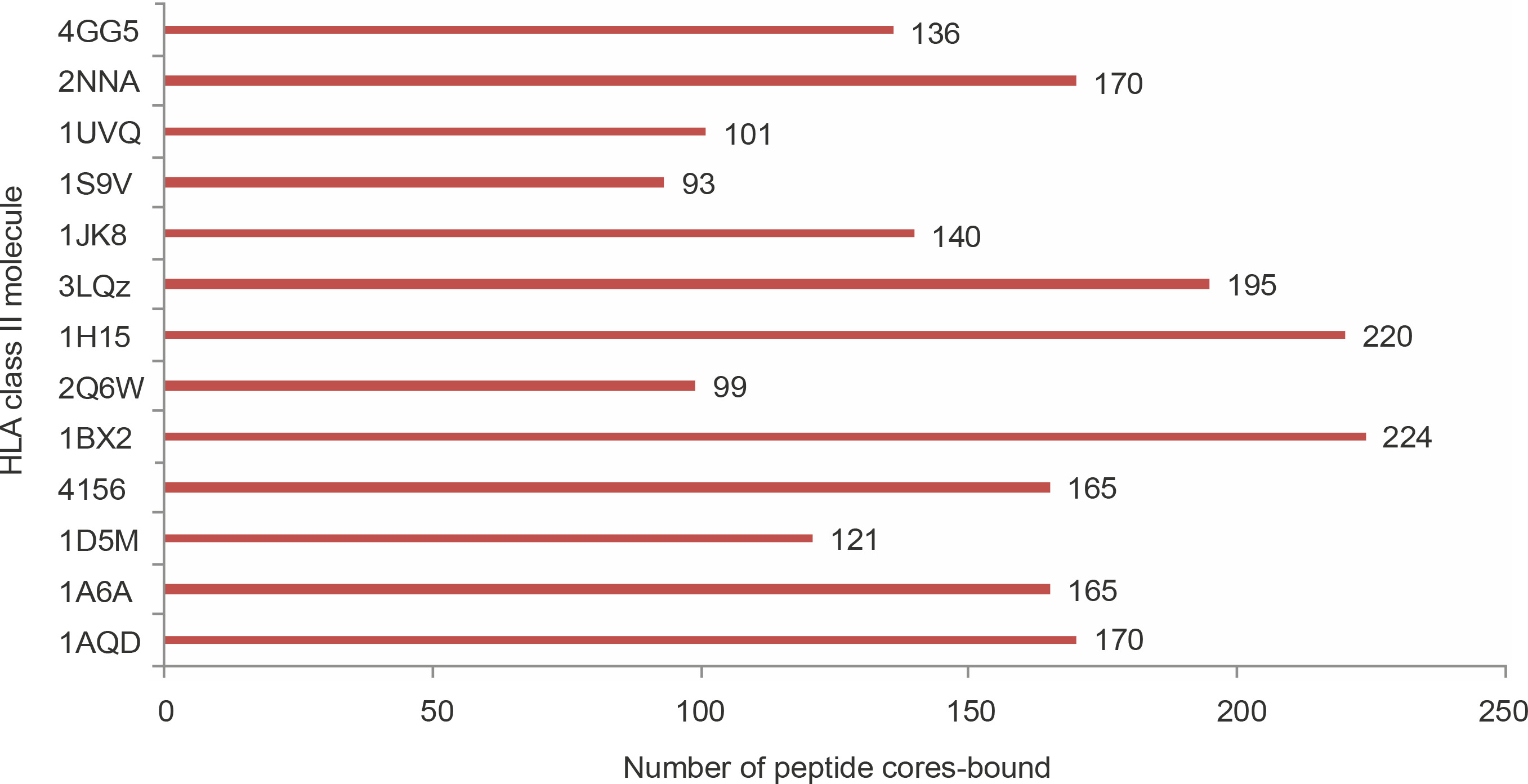
The 545 strong binding non-self peptides contained 308 nonameric core regions. To analyze the structural feasibility of the predicted binding of these nonameric peptides to HLA class II molecules, molecular docking of the peptides to 13 different HLA molecules was performed. Of the 308 nonamers, 289 were shown to bind to the peptide binding groove of at least one HLA molecule with glide score better than –10 kcal/mol (Supplementary materials 2B). This result shows a strong positive correlation between NetMHCII prediction and the possibility of a favorable binding interaction between a predicted peptide and an HLA molecule. Fifteen core peptides bound to all the 13 HLA molecules used in the study with glide scores better than –10 kcal/mol (Table 4). Seventy five core peptides bound to 10 or more of the HLA structures studied. The number of peptide cores bound by the PDB structures varied. Few proteins such as 1BX2 (HLA-DR), 1H15 (HLA-DR), and 3LQZ (HLA-DP) bound to significantly higher number of peptides than proteins such as 1S9V (HLA DQ), 2Q6W (HLA-DR), and 1UVQ (HLA-DQ), as shown in Figure 2. SACOL0907, SACOL0887, and SACOL08581 identified as potential vaccine candidate antigens using NetMHCII prediction were also shown to produce good number of core peptides that bound at least one of the HLA molecules with glide scores better than –10 kcal/mol. Although the bifunctional autolysin (Atl1) SACOL1062 produced 24 core peptides that bound to any one of the HLA molecules with glide score better than –10 kcal/mol, it may not be considered as a good vaccine candidate protein because of its low epitope density (0.03).
Table 4
CD4+ T cell epitopes that docked to all the HLA class II molecules with glide scores better than –10 kcal/mol
Peptide-HLA interactions
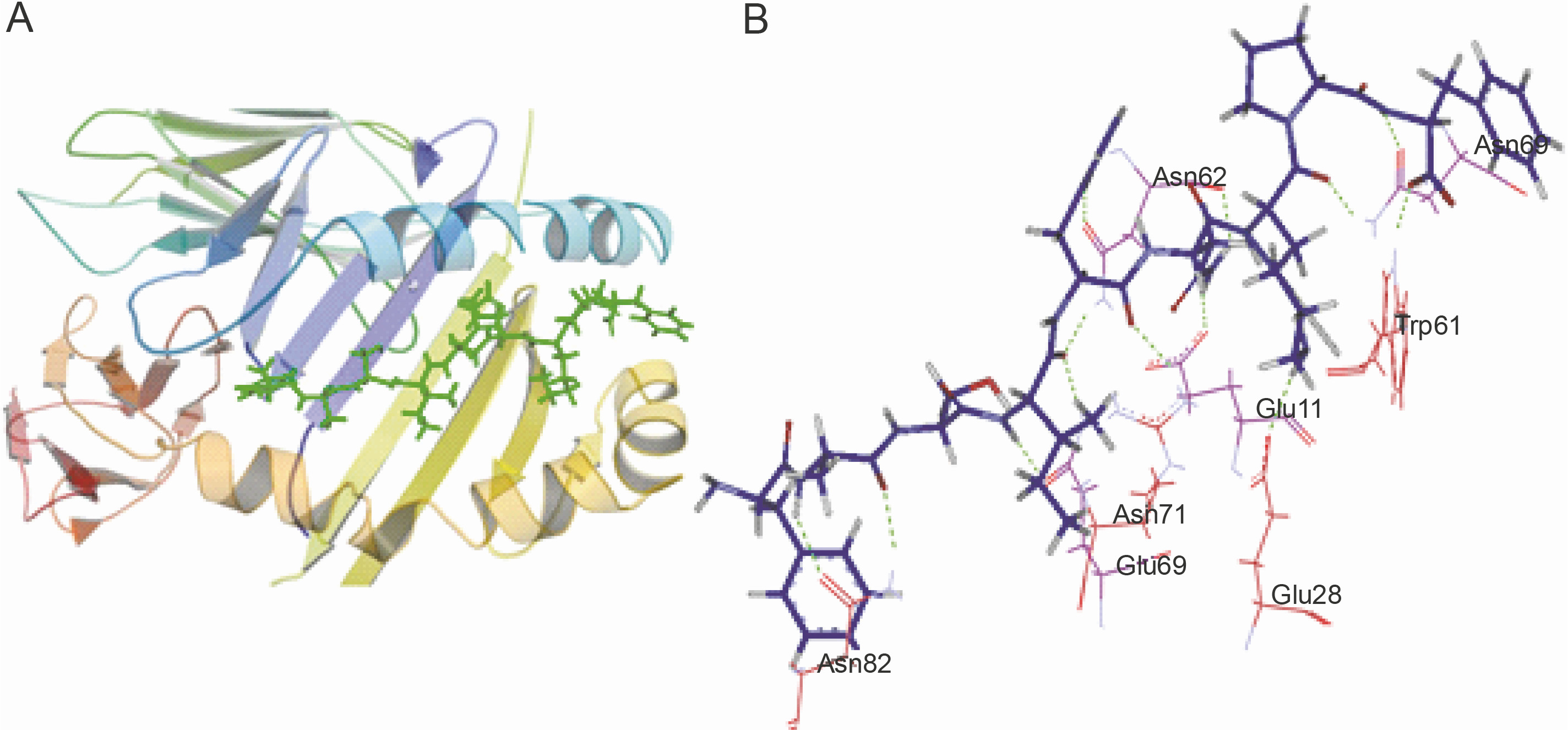
The peptide core sequences that bound with the highest negative glide score to the HLA class II structures used in the study are shown in Table 5. The structure of the predicted non-self peptide-HLA complex was compared to that of the resident peptide-HLA complex. The hydrogen bonding interactions established by the docked top scoring peptide and the resident peptide with the peptide binding groove were also compared. Ten of the 13 top scoring peptides showed better glide scores than the resident peptides (refer to Table 5 for the glide scores). Only 1UVQ and 4GG6 docked peptides with inferior glide scores than the resident peptide. 2NNA docked the peptide with glide score comparable to that of the resident peptide. As expected peptides docked to 1UVQ and 4GG6 were shown to have fewer number of hydrogen bonds than the resident peptides. To illustrate the mode of binding of the peptides to the binding groove of HLA, the docking of the peptide core YASIHNKPF to 1AQD is shown in Figure 3A. YASIHNKPF docked 1AQD with a significantly better glide score than the resident peptide. The hydrogen bonding interactions are shown in Figure 3B.
Table 5
Glide scores of the top-scoring peptides compared to that of resident peptides
Fig. 3
Peptide-HLA interaction. A) the docking of the peptide YASIHNKPF to 1AQD; the peptide is shown as lines and sticks in green; the floor of the peptide binding groove is formed by the β sheets contributed by α and β chains of the protein (shown in blue and yellow, respectively); the floor is bordered by α helices contributed by the protein chains; the peptide docked with a glide score of –21.087 kcal/mol; B) hydrogen bonding interactions established by YASIHNKPF with the peptide binding groove of 1AQD; the peptide is shown as the thick blue line; the green dotted lines represent the hydrogen bonds; the amino acid residues shown in purple are contributed by the α chain of the HLA molecule, while those marked in red are contributed by the β chain
Discussion
Development of a vaccine against S. aureus has remained a formidable challenge, and it still looms large as an unaccomplished task. Research evidence suggests that antigens that activate CD4+ T cells may afford protection against S. aureus infections (Spellberg and Daum, 2012; Proctor, 2012; Giersing et al., 2016; Lee et al., 2020). It is a laborious and time-consuming process to examine each protein of an organism experimentally for the potential to generate CD4+ T cell responses. An initial in silico screening for vaccine candidate antigens would help in limiting the number of proteins to be examined experimentally. Immunoinformatic approaches to design vaccines against S. aureus by focusing on a limited set of antigens have been reported (Delfani et al., 2015; Hajighahramani et al., 2017). In the present study, to identify CD4+ T cell epitopes that could be used in a vaccine formulation against S. aureus, 16,724 peptides were generated from 59 secreted proteins and evaluated for their efficacy to bind HLA class II alleles by using a combination of immunoinformatic prediction and molecular docking. Only 3.25% of the peptides showed strong binding affinity to HLA alleles and were non-self in humans. Importantly, the study showed that only 1.5% of the HLA-binding peptides exhibited significant promiscuity (8 or more alleles) in allele binding. At the maximum, a peptide could bind to only half (50%) of the 26 alleles studied. This suggests that a single protein/epitope strategy for developing S. aureus vaccine would be futile and corroborates the view that vaccine formulations should be composed of multiple antigens/epitopes to achieve wider population coverage (Nezafat et al. 2016; Mahmoodi et al., 2020). In fact, many of the suspended trials to develop a vaccine used single proteins as candidate antigens (Weems et al., 2006; Fowler and Proctor, 2014). An analysis of the degree of conservation of the predicted vaccine candidate epitopes using BLASTp and Constraint-based Multiple Alignment Tool (COBALT) revealed that the epitopes are well conserved across different strains of S. aureus. Hence, the peptides may be assumed to be effective against diverse strains of the bacteria.
Most of the studies used to identify and validate vaccine candidates against S. aureus employ mice and other animals as models. The restricted binding of peptide epitopes in a sequence-dependent manner to HLA class II molecules suggests that the results obtained from animal models cannot be correlated to human T cell response. It is quite unlikely that a peptide binding to mice MHC would bind to human HLA class II molecules (Kumánovics et al., 2003; Lawrence et al., 2012, Salgado-Pabón and Schlievert, 2014). Moreover, in vivo studies have limitations in identifying promiscuous peptides from proteins because of the relatively low number of allele combinations that can be tested. Computational approaches enable to rapidly identify peptides that can bind to a high number of HLA alleles because the sequence/structure data of many HLA alleles are available in public databases. The human HLA class II restriction patterns in proteins can be conveniently identified using in silico approaches.
Apart from simply relying on a prediction algorithm to identify epitopes, we further studied the structural feasibility of the binding of peptides to HLA class II molecules using molecular docking. In general, the results of the docking studies correlated well with the binding predictions. The peptide-HLA binding interactions for the top-scoring peptides obtained in the docking studies were stabilized through hydrogen bonding interactions.
Although it is well understood that a screening attempt exclusively relying on computational tools cannot be viewed with full confidence considering the complex nature of immune recognition, it is noteworthy that a few proteins that emerged as potential vaccine candidates in this study were earlier experimentally determined to be immunogenic. Among the peptides identified as vaccine candidates, NYFRFQYFNPLKSER and RFQYFNPLKSE RYYR were predicted to bind to 13 and 8 alleles, respectively, and are generated by the putative protein SdrH. This surface adhesin belonging to a protein multigene family was experimentally found to induce high epitope-specific anti-staphylococcal antibody titers in both healthy and infected individuals (Dryla et al., 2005). The same study also identified Empbp1, another protein predicted to harbor promiscuous peptides in the present study, as capable of inducing epitope-specific antibody responses. Other members of this family were found to afford protection against invasive diseases or a lethal challenge with human clinical S. aureus isolates in murine models (Stranger-Jones et al., 2006; Luna et al. 2019). These experimental data increase the confidence on the predictions made in the study. Another noteworthy peptide epitope identified as a promising epitope vaccine candidate is KFDQSKYLMMYNDNK harbored by the superantigen Seb. Seb is known to be resistant to protease cleavage, and unlike normal antigens, it binds directly to MHC-II molecules, causing a cytokine storm that leads to toxic shock syndrome (Pinchuk et al., 2010). However, modified forms of the protein lacking superantigenicity are known to induce protective immune responses, and STEBVax, a candidate vaccine based on a modified Seb, has completed phase I clinical trials (Hudson et al., 2013; Chen et al., 2016; Choi et al., 2017). The CD4+ T cell epitope identified in the present study will be devoid of super antigenicity as it contains only a part of the protein. Since the peptide has shown potential to bind to MHC class-II molecules with high promiscuity it holds potential to be developed as a potent epitope vaccine.
Although the major focus of this study was to identify peptide epitopes, few proteins also emerged as potential vaccine candidates. Among the proteins identified as CD4+ T cell antigens based on epitope density are staphylococcal enterotoxin type I (Sei), Seb, and Empbp1. Staphylococcal enterotoxins are superantigenic toxins produced by S. aureus. As already discussed, mutant or modified Seb devoid of antigenicity are well-studied vaccine candidates. Empbp1 is a protein that interacts with fibronectin, fibrinogen, and vitronectin and plays a key role in the formation of abscesses (Crosby et al., 2016). Thus, it is a key virulence factor in S. aureus. The fact that Empbp1 was earlier identified as an in vivo-expressed vaccine candidate in S. aureus (Etz et al., 2002) further adds confidence to the predictions made in this study. The epitopes and antigens predicted to trigger a CD4+ T cell response in the present study can be included in a multiepitope/multivalent vaccine that can induce cell-mediated immunity along with humoral immunity and lead to the formulation of a well-balanced vaccine cocktail against S. aureus.
Conclusion
In the present study, a combined immunoinformatic and molecular docking approach was used to screen the secretome of S. aureus to identify CD4+ T cell epitopes with attributes of potential vaccine candidates. A total of 545 potential non-self epitopes were identified and validated using molecular docking. The list was further refined based on allele coverage, a key element in rational vaccine design. Eleven peptides containing nine unique peptide cores were identified as potential vaccine candidates. A few proteins that harbor HLA-restricted epitopes with high density were also identified. Apart from using a prediction algorithm to identify epitopes, the structural feasibility of the binding of the peptides to HLA class II molecules was further studied using molecular docking. The molecular docking studies validated the prediction results as the peptides interacted favorably with the peptide binding groove of the crystal structures of HLA class II molecules. It is, however, well understood that a screening attempt exclusively relying on computational tools cannot be viewed with full confidence considering the complex nature of immune recognition. Therefore, to translate the in silico results into a clinically viable vaccine, the peptides and proteins predicted as candidate antigens should be tested in vitro and in vivo singly or in combination with other antigens.